
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Viedma ripening: a reliable crystallisation method to reach single chirality
Leyla-Cann
Sögütoglu
,
René R. E.
Steendam
,
Hugo
Meekes
,
Elias
Vlieg
* and
Floris P. J. T.
Rutjes
*
Radboud University, Institute for Molecules and Materials, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: F. Rutjes@science.ru.nl; Fax: +31 24 365 3393; Tel: +31 24 365 3202
First published on 13th July 2015
Abstract
Crystallisation processes have evolved to practical methods that allow isolation of an enantiopure product in high yield. Viedma ripening in particular enables access to enantiopure products in a reliable way, simply through grinding of crystals in a solution. This tutorial review covers the basic principles behind asymmetric crystallisation processes, with an emphasis on Viedma ripening, and shows that to date many novel organic molecules can be obtained in enantiopure solid form.
 Leyla-Cann Sögütoglu | Leyla-Cann Sögütoglu (1990, Eindhoven, NL) is a chemistry student at the Radboud University in Nijmegen, where she obtained her BSc in 2013. As a Master's student she performed research at the interface of solid state chemistry and solid state NMR under supervision of Dr H. Meekes and Dr E. van Eck, focussing on the crystal structure of hydrophobic amino acids. In a second Master's research project, she currently studies the biomimetic synthesis of magnetite in the biomineralisation group of Prof. N. Sommerdijk at the Eindhoven University of Technology. |
 René R. E. Steendam | René Steendam (1986, Hardenberg, NL) studied chemistry at the Radboud University in Nijmegen. His PhD research took place in the groups of Prof. E. Vlieg and Prof. F. Rutjes and focussed on merging organic synthesis with Viedma ripening. He recently was awarded an NWO Rubicon grant to continue investigating the Viedma ripening process as a post-doc in the group of Prof. J. ter Horst at the University of Strathclyde (UK). |
 Hugo Meekes | Dr Hugo Meekes (1959, Lichtenvoorde, NL) received his PhD at the University of Nijmegen (NL) under supervision of Prof. A.G.M. Janner and Prof. P. Wyder in 1988. In 1987 he became assistant professor at the University of Nijmegen (nowadays Radboud University) in the Solid State Chemistry group. His research interests include experimental studies and automation of the prediction of morphology of crystals as well as of polymorphism in crystals, modelling of nucleation of polymorphic forms, polymorph prediction, deracimization of chiral molecules using crystallization. He is currently topic editor of the ACS-journal Crystal Growth & Design and board member of the Dutch Association for Crystal Growth. |
 Elias Vlieg | Prof. Elias Vlieg (1961, Leeuwarden, NL) obtained his PhD in physics in 1988 at the University of Leiden on work performed with Prof. J. F. van der Veen at the FOM institute AMOLF in Amsterdam. After a two-year post-doc at AT&T Bell Laboratories (USA) he returned to AMOLF as a group leader. In 1998 he became full professor in Solid State Chemistry at the Radboud University in Nijmegen. His research interest focuses on crystal growth and includes the topics chiral separation, protein crystallization, self-assembly of monolayers, solar cells and the structure of solid–liquid interfaces. He is director of the Institute for Molecules and Materials (IMM) and co-founder and director of the spin-off company tf2 devices. |
 Floris P. J. T. Rutjes | Prof. Floris Rutjes (1966, Heiloo, NL) received his PhD at the University of Amsterdam (NL) under supervision of Prof. Nico Speckamp in 1993. He then was post-doc with Prof. K.C. Nicolaou (The Scripps Research Institute, La Jolla, USA) completing a total synthesis of brevetoxin B. In 1995, he became assistant professor at the University of Amsterdam and in 1999 full professor in synthetic organic chemistry at Radboud University in Nijmegen. His research interests include the application of asymmetric catalysis in the synthesis of biologically relevant molecules, and flow chemistry. He is co-founder of the spin-off companies Chiralix and FutureChemistry and director of the Educational Institute for Molecular Sciences. |
Key learning pointsSolid-state versus molecular chiralityAsymmetric crystallisation from solution Deracemisation through Viedma ripening Crystal growth Racemisation |
(1) Introduction
Mirror symmetry is widely present throughout Nature and has fascinated and inspired mankind to date. Although any object has a mirror image, not every mirror image is identical to the original. Such objects are coined “chiral”, derived from the ancient Greek word χειρ (kheir) which means “hand”. Chirality is also present at the molecular level: a molecule is chiral – and consists of two mirror-image isomers or enantiomers – if it has no internal plane of symmetry. Such a situation occurs for example if a molecule contains a tetrahedral carbon atom bearing four different substituents. Thermodynamically, the enantiomers (denoted as S or R) are equally likely to exist, yet most chiral molecules in living organisms are overrepresented in one enantiomeric form. How this molecular asymmetry emerged under prebiotic conditions remains a much studied topic in science.1 The single handedness in living systems furthermore implies that the two enantiomers of a molecule interact differently with chiral biomolecular receptors. This has enormous consequences for the pharmaceutical industry as generally only one of the two enantiomers exerts the desired medicinal effect. Due to the effect that active pharmaceutical ingredients tend to become more selective with increasing complexity, progressively more single enantiomeric drugs are being launched as compared to racemic- or achiral drugs.2 Therefore, strategies to create single enantiomeric compounds are of pivotal importance to the fine chemical and pharmaceutical industry. Over the past ten years, amongst a variety of chemical and enzymatic methods, Viedma ripening (Fig. 1) has been established as a reliable solid-state method for the deracemisation of racemic mixtures of crystalline compounds into single enantiomers, simply by continuously grinding a suspension.3 The process is not yet used on an industrial scale.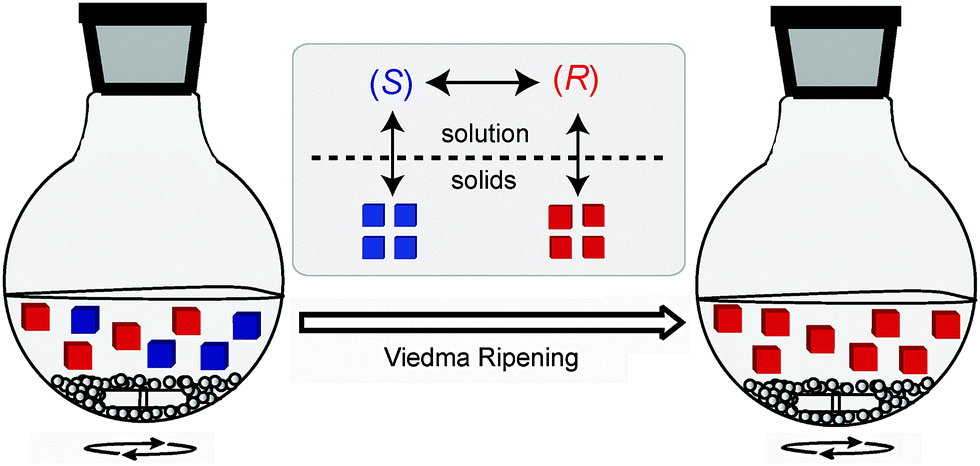 | ||
| Fig. 1 Schematic representation of Viedma ripening. | ||
After its discovery in 2005, an increasing number of reports demonstrate the broad applicability of Viedma ripening which enabled the complete deracemisation of a large variety of crystalline chiral compounds.3 Section 2 of this tutorial review covers the basic principles behind asymmetric crystallisation processes with a brief history on the discovery of Viedma ripening. The principles underlying Viedma ripening are addressed in Section 3. Finally, a number of recent applications involving crystallisation-induced deracemisation methods are highlighted in Section 4.
(2) Asymmetric crystallisation
Nucleation
The very first aggregate of growth units (molecules, ions, atoms) large enough to grow to a macroscopic crystal, is called a nucleus. Nucleation in solution starts when the compound is not soluble any more. Common salt (NaCl) for example, will become insoluble in water (i.e. the solution becomes saturated) at room temperature at a concentration of 357 g L−1 and will start to nucleate if the solubility is decreased further, typically by cooling or through evaporation of the solvent. The solution or liquid from which crystals precipitate is called the mother liquor. Two types of nucleation are generally distinguished, called primary and secondary nucleation (Fig. 2). Primary nucleation is the initial formation of a crystal when there are no other crystals present. Secondary nucleation is the formation of nuclei from an existing macroscopic crystal when this crystal is crushed into smaller pieces, for example as the result of convection,4 crystal–crystal collisions or mechanical breakage by a stirrer bar. Primary nucleation can proceed homogeneously or heterogeneously. The nucleation is called homogeneous in case the compound nucleates without interplay of any media other than the solution. The term heterogeneous nucleation, on the other hand, is used when the compound nucleates onto another compound or dust particles in the solution. Crystal growth starts nearly always via heterogeneous nucleation.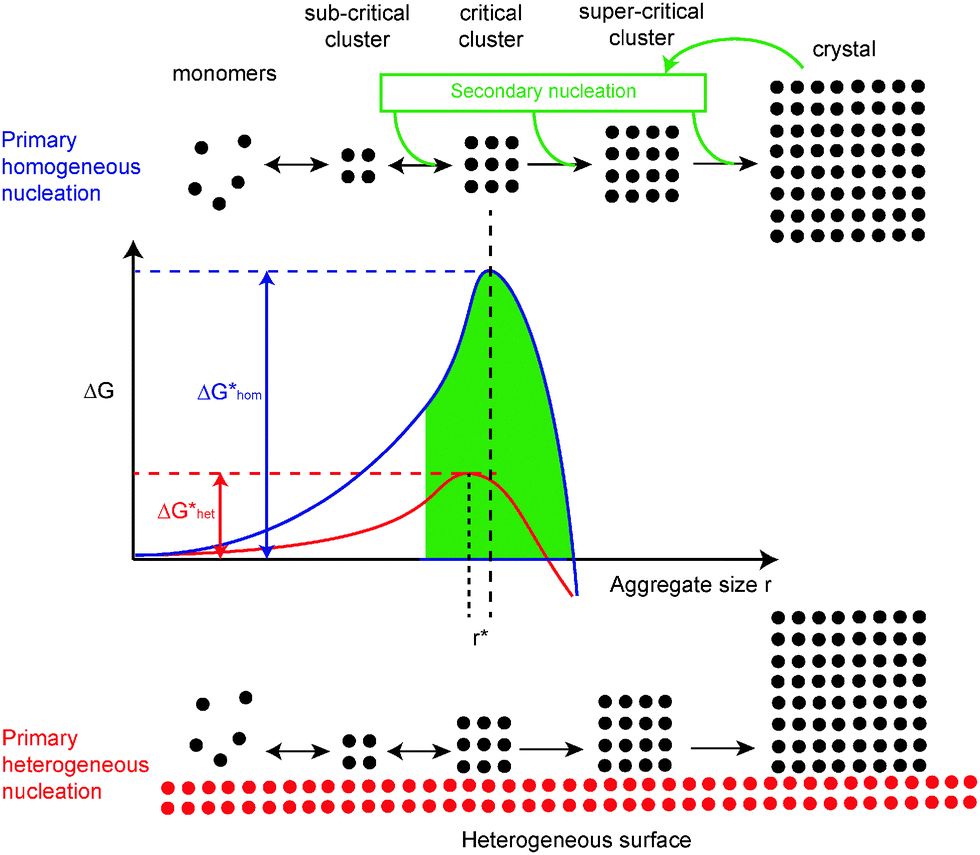 | ||
| Fig. 2 The formation of crystals from solution proceeds either through primary-(homogeneous or heterogeneous) or through secondary nucleation. | ||
In a simplified view, crystal growth encounters a barrier as a result of unfavourable crystal-solution surface energy versus favourable bulk energies. The critical nucleus size (r*) is reached when the favourable and unfavourable energy contributions just counterbalance. Note that the critical nucleus size for primary and secondary nucleation does not need to be the same as it depends on an interfacial energy contribution. Once an aggregate is larger than the critical nucleus it can grow ‘downhill’ spontaneously. In other words, the crystal as thermodynamic end product has a kinetic barrier called nucleation barrier. The barrier can be reduced by primary heterogeneous nucleation (red curve) or even ‘disregarded’ by using secondary nucleation (green area), which supplies clusters, usually larger than the critical nucleus.
For further reading on the crystallisation of molecules from solution, the reader is advised to consult a previously reported tutorial review.5
Crystallisation of enantiomers
Chiral discrimination and enantiomeric separation is extremely difficult to achieve in solution without chiral reagents. The synthesis of chiral molecules typically results in a racemic mixture in which both enantiomers are present in approximately equal amounts. The enantiomeric excess (ee) is used to define the ratio between enantiomers:| ee (%) = (([R] − [S])/([R] + [S])) × 100 | (1) |
In the solid state, chiral discrimination is easier to realise. This especially applies to molecules which, in the crystal structure, have a greater affinity for the same enantiomer than for the opposite enantiomer (Fig. 3). Such a racemic conglomerate is in fact a mechanical mixture of enantiomerically pure crystals of one enantiomer and its opposite. This is the case for approximately 5–10% of all chiral crystalline molecules.6 When molecules have a greater affinity for the opposite enantiomer than for the same enantiomer, the molecules form a single crystalline phase in which the two enantiomers are present in an ordered 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The crystallographic unit cell contains both enantiomers and the solid is called a racemic compound or true racemate. Approximately 90–95% of all crystalline racemic mixtures form racemic compounds. Finally, in less than 1% of the cases a racemic mixture crystallises as a solid solution, containing molecules of each enantiomer in a random arrangement. A unit cell can still be assigned, but the molecular filling of this unit cell is not as well-defined as in the case of a true crystal.
:1 ratio. The crystallographic unit cell contains both enantiomers and the solid is called a racemic compound or true racemate. Approximately 90–95% of all crystalline racemic mixtures form racemic compounds. Finally, in less than 1% of the cases a racemic mixture crystallises as a solid solution, containing molecules of each enantiomer in a random arrangement. A unit cell can still be assigned, but the molecular filling of this unit cell is not as well-defined as in the case of a true crystal.
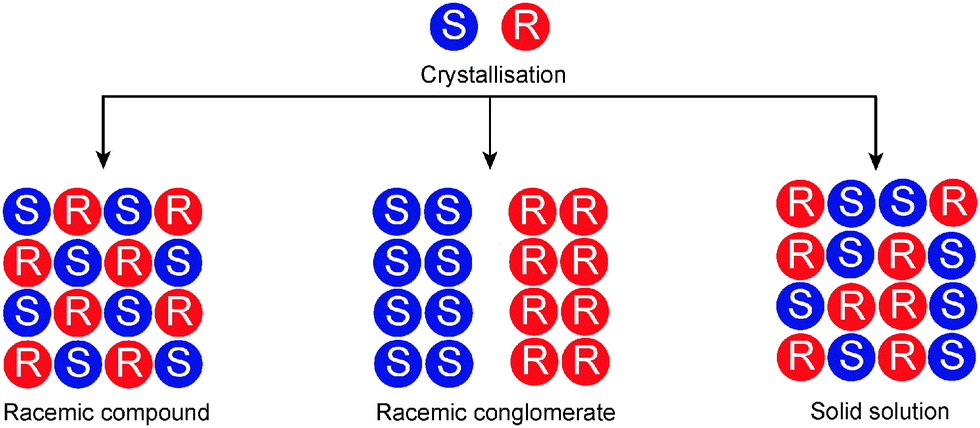 | ||
| Fig. 3 Enantiomers crystallise either as a racemic compound, racemic conglomerate or a solid solution. | ||
Total spontaneous resolution
Chiral discrimination in the solid state can only be achieved with molecules that form racemic conglomerate crystals. Nucleation of these molecules from a homogeneous solution generally proceeds to give a crystal of single chirality (Fig. 4a). In this way, the solution becomes enriched in the opposite enantiomer that will also crystallise upon further increase of the supersaturation. Eventually, the overall solid state is racemic yet each crystal is enantiopure. If the crystals can be separated from each other, one can obtain enantiopure products and in this way perform spontaneous resolution. Louis Pasteur obtained enantiopure conglomerate crystals through the manual separation of crystals of sodium ammonium tartrate.7 Although Pasteur was able to separate the crystals by hand based on morphological chirality, many crystals do not display such a chiral morphology. This separation method is still being used, albeit in a more practical way.8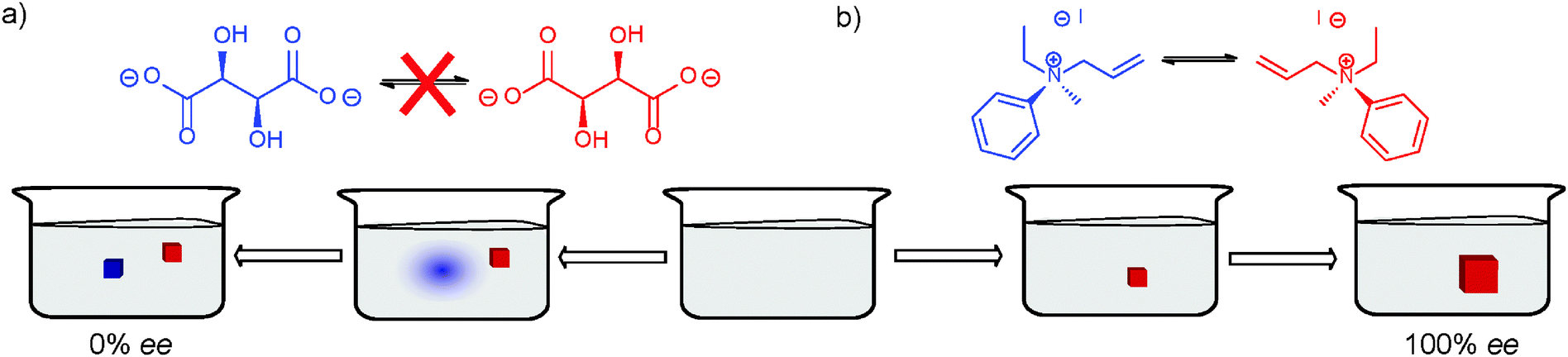 | ||
| Fig. 4 Crystallisation of conglomerate-forming molecules in (a) the absence and (b) presence of solution-phase racemisation. | ||
To prevent the nucleation of the other enantiomer, Egbert Havinga used conglomerate-forming quaternary ammonium iodide (Fig. 4b) that racemises in solution as a spontaneous process toward maximal entropy.9,10 Because of the decrease in supersaturation due to the first crystal formed (Fig. 2), primary nucleation of either enantiomer will less likely take place. As the mother crystal grows larger, it retains its chirality by taking up only the monomers with matching chirality. The supply of these monomers is maintained through racemisation in solution. This way enantiopure crystals spontaneously form without the need for separation. This process is also known as total spontaneous resolution.6
Havinga postulated in 1941, at the Dutch organic chemistry conference, the three requirements for a racemic mixture to have a high probability to undergo total spontaneous resolution:9
(1) The compound must form separate R and S crystals (i.e. the compound is a racemic conglomerate).
(2) The compound must be able to racemise in solution, possibly aided by a catalyst speeding up the racemisation.
(3) The rate of crystal nucleation is low, while the rate of crystal growth is high. The rate of racemisation should be high as well.
At first Havinga was unaware of the impact of his research, which was published in the Chemisch Weekblad, a journal written in Dutch. Soon after a publication of a model on spontaneous asymmetric synthesis by the theoretical physicist Sir Charles Frank,11 Havinga published his experimental findings again, but this time in English in an international journal.10 Interestingly, an often overlooked detail in the experimental section of the Dutch paper is that Havinga mentions that in some experiments several crystals were formed as the result of agitation. When combined, the mixture of crystals still displayed optical activity. Havinga proposed that these crystals were grown from seeds originating from the initial mother crystal (secondary nucleation) and therefore were of the same handedness (Fig. 5).
 | ||
| Fig. 5 Primary nucleation to give an enantiopure crystal which can grow larger (top) or will undergo secondary nucleation (bottom). | ||
This effect of secondary nucleation was studied in 1990 in more detail by Kondepudi et al. who found that crystallisation from a stirred solution12 or melt13,14 leads to many small crystals which are nearly all of the same handedness. In their solution-phase experiments, the compound used was sodium chlorate which is achiral as a molecule in solution but in the solid state the arrangement is such that the crystals become chiral (Fig. 6).
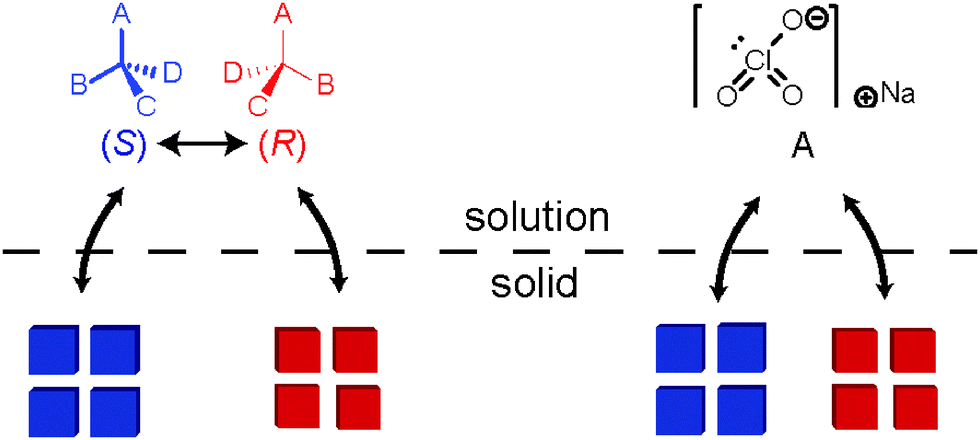 | ||
| Fig. 6 Solution-solid equilibria of a chiral molecule (left) and sodium chlorate (right). | ||
Kondepudi et al. showed that nearly all crystals (99.7% of all crystals formed in an experiment) had the same chirality when an aqueous solution of sodium chlorate was stirred during crystallisation. He stated that “in order to produce total asymmetry of close to 100% in the product crystals in every try, autocatalysis and competition between the L- and D-crystals are needed.” In Kondepudi's experiment too, all requirements for total spontaneous resolution as stated by Havinga are met: (1) NaClO3 crystallises as chiral crystals (thus being a conglomerate in the solid state), (2) racemisation in solution is not even needed, because the building block is achiral and (3) the rate of primary nucleation was kept low, while the rate of crystal growth was high. The latter condition was possible, because Kondepudi made use of the rapid generation of secondary nuclei which reduced the concentration to a level at which the rate of primary nucleation is virtually zero under stirring conditions. Primary nucleation is a cumbersome process, with a kinetic barrier of reaching the critical nucleus, whereas secondary nuclei larger than the critical nucleus grow as soon as they are formed. As such, the left- and right-handed nuclei generated through secondary nucleation compete for the solute and the rapid crystal growth suppresses primary nucleation and therefore the formation of nuclei of the opposite handedness. In addition, Kondepudi noticed that when the solution is not stirred, there is no preference for one chiral form over the other. This means that all of the nuclei are produced through primary nucleation, homogeneous or heterogeneous, and their handedness is at random. In this case too, the depletion of the solute due to crystal growth may eventually stop the primary nucleation, but no chiral resolution occurred in these trials.
Later, Durand et al. showed that the asymmetry obtained through stirring-induced crystallisation of a conglomerate-forming achiral molecule can be converted to molecular chirality.15 Stirring during the crystallisation of conglomerate-forming molecules which racemise in solution provides an attractive route to obtain molecules in high ee and high yield.16 However, it is not the most reliable approach in reaching an enantiopure product as nucleation of the unwanted enantiomer still can take place.
(3) Viedma ripening
In an attempt to test the hypothesis of secondary nucleation, Cristobal Viedma repeated the Kondepudi experiments in 2004, but under conditions which facilitate a much higher nucleation rate leading to many primary nucleation events of sodium chlorate.17 Intriguingly, the solid state was still found to be of single chirality despite the fact that the high nucleation rate led to the nucleation of both enantiomers. In a follow-up experiment, Viedma prepared a saturated solution already containing crystals of both chiral forms which he then subjected to intensive grinding using glass beads in a closed system under isothermal conditions (Fig. 7).18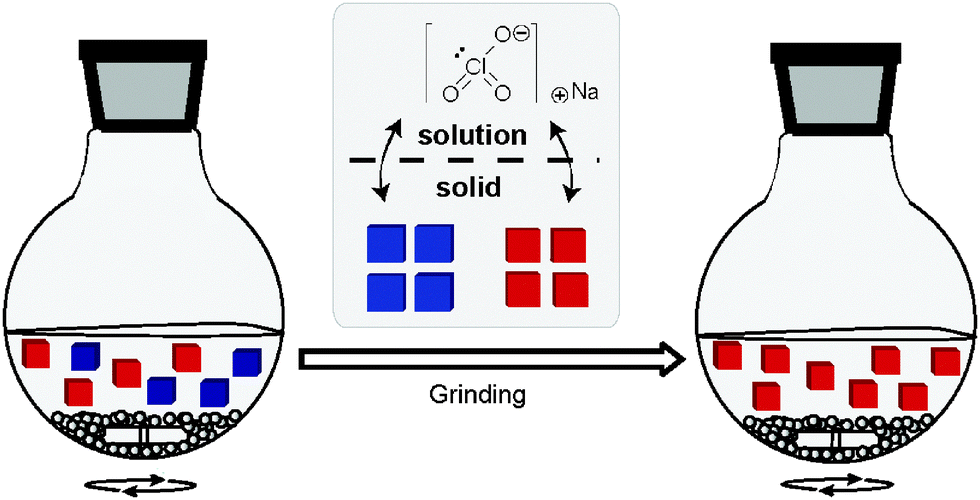 | ||
| Fig. 7 Schematic representation of Viedma's experiment. | ||
Viedma discovered that the initially racemic mixture of sodium chlorate crystals was, over a period of several days, completely transformed into an end state in which all of the crystals were of one chiral form. This transformation, which is now called Viedma ripening, involves solid-to-solid deracemisation instead of solution-to-solid deracemisation and is therefore significantly different as compared to total spontaneous resolution.
Like Kondepudi et al., Viedma used the achiral molecule sodium chlorate in his experiments. In 2008, Noorduin et al. extended Viedma ripening to intrinsically chiral molecules.19 They showed that amino acid derivatives, such as the one depicted in Fig. 8, can also undergo solid-to-solid deracemisation. These molecules also form conglomerate crystals and undergo racemisation in solution using a strong base. Noorduin et al. showed that Viedma ripening of this compound can readily be scaled up to larger volumes of up to 320 mL of solvent, thereby demonstrating the industrial viability of Viedma ripening.20 In addition, other pharmaceutically-relevant molecules, such as Naproxen21 and a Clopidogrel intermediate,22 were found to undergo complete deracemisation through Viedma ripening. The final configuration of the product can easily be controlled using additives,19 difference in crystal size between the enantiomers23 and even the order of process steps.24
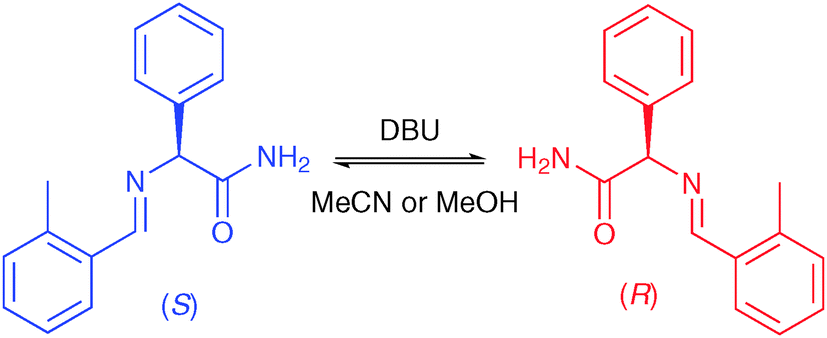 | ||
| Fig. 8 Extension of Viedma ripening to intrinsically chiral molecules that form racemic conglomerate crystals. | ||
During the past decade, several research groups have studied the mechanism behind this intriguing transformation. Although “the practical execution of the process is remarkably simple”25 the underlying mechanism is more complicated. After symmetry breaking of the racemic mixture of crystals due to random local fluctuations in ee or local fluctuations in the crystal size distribution (CSD) difference between the enantiomers, the system undergoes complete solid state deracemisation through an autocatalytic feedback mechanism in which the initial ee is amplified exponentially to an enantiopure end state. To date, many computational studies have been carried out to explain the mechanism behind Viedma ripening. To account for all these models, however, is beyond the scope of this review. Still, most modeling studies describe the Viedma ripening process along the four factors described in the following paragraphs (also indicated with the numbers in Fig. 9).25
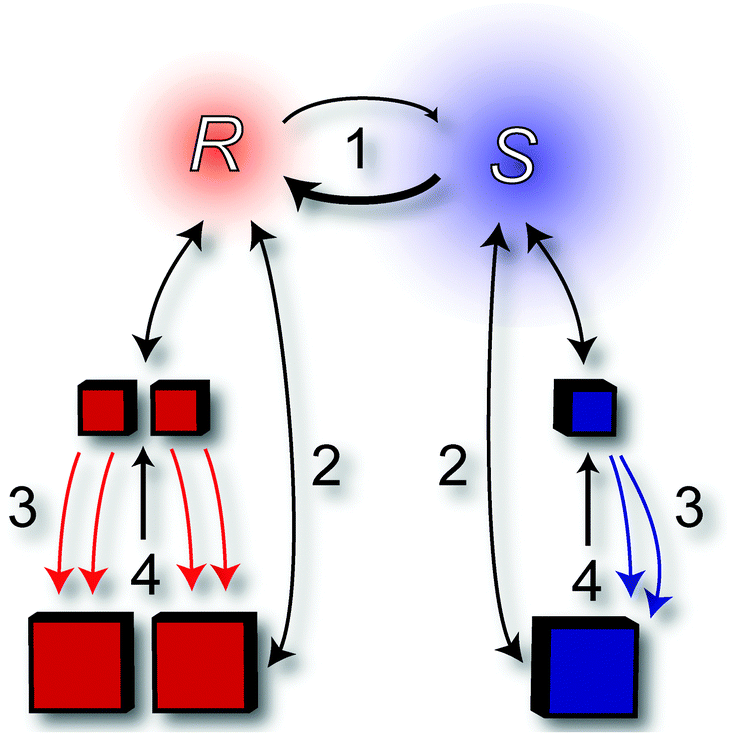 | ||
| Fig. 9 Schematic representation of a proposed mechanism behind Viedma ripening. The numbers are explained in the main text. | ||
1. Racemisation in solution (which is not needed for achiral molecules that crystallise as a racemic conglomerate)
In order to convert all enantiomers in the solid phase to one form, the molecule must continuously racemise in solution. Furthermore, chiral clusters of the (S)-enantiomer have a lower probability to encounter a crystal of the same handedness, and thus dissolve more frequently. As a result, the solution becomes enriched with the enantiomer that is opposite to the one that is enriched in the solid phase. The enantiomer that is enriched in solution undergoes racemisation leading to the supply of more of the enantiomer that corresponds to the one that is enriched in the solid phase.2. Ostwald ripening
Ostwald ripening, first described by Wilhelm Ostwald in 1896, is the continued dissolution and growth of crystals to reach a thermodynamically stable state wherein the surface area to volume ratio of the solid system is minimised. That is, larger crystals grow at the expense of smaller crystals. The latter is implicitly shown in Fig. 2, as the extrapolated Gibbs free energy is still lowering for larger particle size. There is a tendency towards the largest aggregate or crystal. When crystals of one enantiomer are sufficiently larger than the other, Viedma ripening leads to complete deracemisation in which the final product corresponds to the initially larger crystals (survival of the fittest).233. Enantioselective incorporation of clusters into larger crystals26
Fig. 9 shows the incorporation of both solute molecules as well as clusters into larger crystals of the same handedness. During Viedma ripening, crystals are continuously broken into clusters. The reincorporation of these clusters into larger crystallites occurs more often for the enantiomer which is in excess in the solid phase. This process, together with attrition, leads to the autocatalytic amplification effect typical for Viedma ripening. Experimental proof for enantioselective incorporation of chiral clusters remains to be found, although some indirect evidence relating to NaClO3, the archetypical compound for Viedma ripening, has been reported.27,284. Attrition
While enantiopure clusters and molecules of R and S incorporate enantioselectively in the bulk crystals R and S respectively, the bulk crystals are ground, producing chiral fragments as well as monomers. The steady attrition also maintains overall small crystal sizes which in turn enhance the Ostwald ripening effect. A higher attrition intensity leads to shorter deracemisation times.29As the following section will show, novel applications involving Viedma ripening and total spontaneous resolution are still being discovered.
(4) Recent examples
This section highlights some examples in recent literature in which an enantiopure product can be obtained either through total spontaneous resolution or Viedma ripening. Noorduin et al. were able to apply Viedma ripening to the deracemisation of an amino acid derivative (Fig. 8) in which the final configuration of the product could be controlled using the rotation sense of circularly polarised light (CPL).30 After the solution was subjected to r-CPL radiation, Viedma ripening reproducibly led to an enantiopure product with the (R)-configuration. An enantiopure product with the (S)-configuration could be obtained when l-CPL was used. The CPL radiation induces the formation of an enantioenriched (i.e. >0% ee) mixture of side products which stereoselectively hamper the crystal growth of one enantiomer.Another contribution to the discussion on the origin of single chirality was proposed by Frank, already in 1953. Frank envisaged that an enantiopure product can in principle be formed from achiral starting materials provided that the product enantiomer catalyses its own formation and at the same time suppresses the formation of the other enantiomer.11 He concluded his article with the sentence “a laboratory demonstration is not necessarily impossible”. It took nevertheless more than 40 years until Frank's concept of asymmetric autocatalysis was experimentally realised. Asakura et al. found that an optically active cobalt complex can be formed through asymmetric autocatalysis.31 In the same year, Soai et al. showed that an initial small amount of chiral product can be amplified to single chirality.32 Without the addition of enantioenriched material from the beginning, however, enantiopure products could not be obtained.33 These results underline the fact that the synthesis of an enantiopure product from achiral reactants without pre-existing enantioenrichment still is extremely difficult to achieve in solution. More recently, some of the present authors successfully extended Frank's concept to a synthetic organic construction reaction34 in which crystals of the product were used as the asymmetric autocatalytic driving force (Fig. 10).35
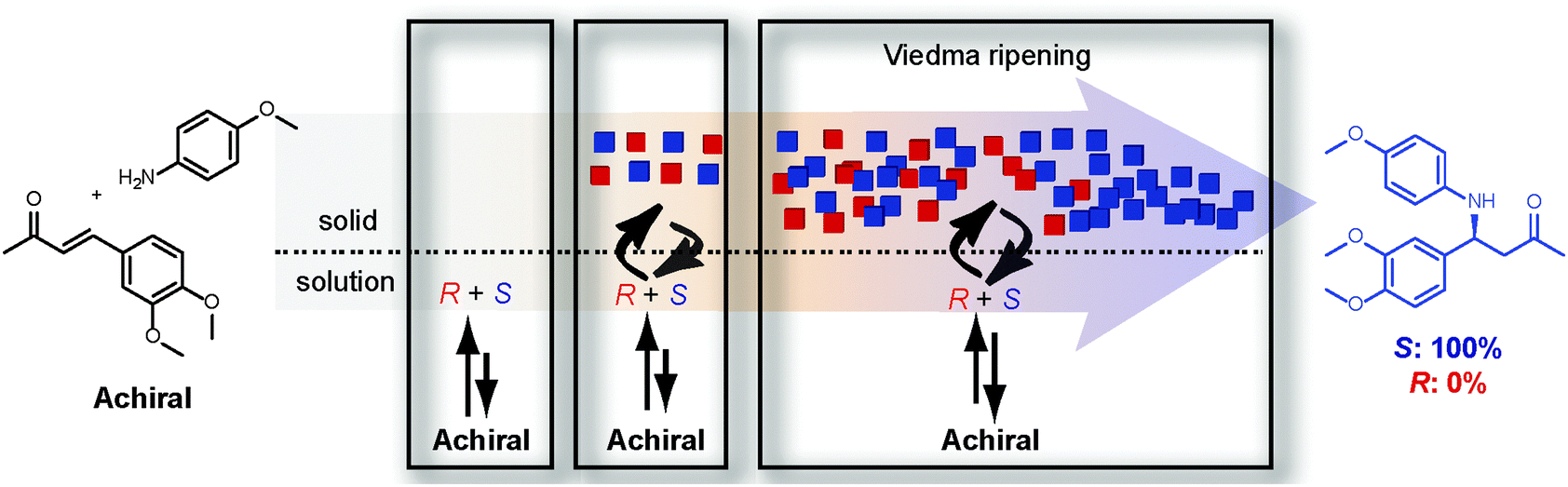 | ||
| Fig. 10 The synthesis of an enantiopure product from achiral conditions. | ||
It was found that the product amine forms racemic conglomerate crystals while in solution it can racemise through a reversible aza-Michael reaction. Starting at a high concentration of achiral reactants, both enantiomers of the product are rapidly formed using DBU as an achiral catalyst in solution. Due to the poor solubility, both enantiomers of the product crystallise to give a crystal-solution system. The latter system subsequently undergoes deracemisation through Viedma ripening due to the applied grinding conditions. This way, an enantiopure product was reproducibly obtained in high yield from achiral reactants in a single reaction. The enantiopure product was formed either in the (R)- or (S)-configuration.
The reversible reaction can also be used to enhance the ee of other conglomerate-forming molecules, as was shown for a reversible Mannich reaction36 and a reversible aldol reaction.37 In addition to a reversible reaction, racemisation can also be induced in different ways as some recent reports have shown (Fig. 11). The group of Hakånsson significantly extended the list of metal complexes that can be resolved through total spontaneous resolution (Fig. 11a).38,39 These complexes have achiral ligands but are chiral (denoted Δ or Λ instead of R and S) as a complex. On the other hand, pyrimidine-derivatives studied by Yagishita et al. exhibit axial chirality and these molecules as such were found to undergo racemisation through an achiral transition state at elevated temperatures without the need for a catalyst (Fig. 11b).40 The combination of seeding and stirring gave the product with an ee of up to 91% through total spontaneous resolution. Another class of compounds that can be obtained in enantioenriched form through total spontaneous resolution are isoindolinones (Fig. 11c). These compounds undergo rapid racemisation through a ring-opened achiral intermediate in the presence of a strong base (DBU). Starting from a clear solution, evaporation of solvent led to the crystallisation of enantioenriched isoindolinones in quantitative yields through total spontaneous resolution.41 Some of the present authors later reported that the isoindolinones can be deracemised completely through Viedma ripening provided that (1) there is sufficient attrition, (2) a suitable solvent is used in which the compounds have a high enough solubility and (3) that racemization is avoided during the ee analysis.42 Moreover, the reported isoindolinones readily racemise in ethanol without a catalyst and this enabled the complete deracemisation of these isoindolinones through Viedma ripening in ethanol.
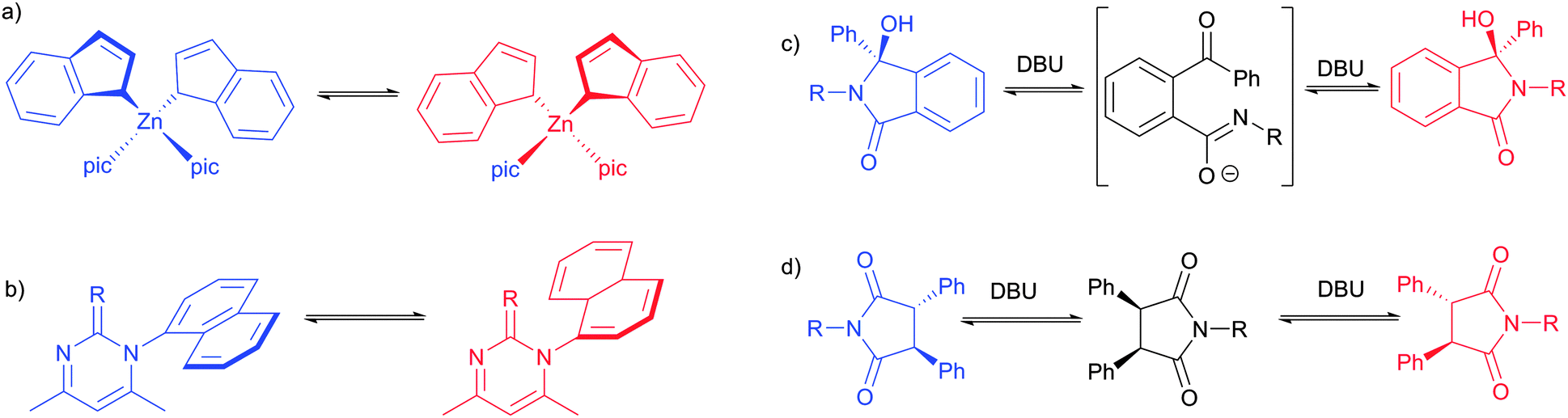 | ||
| Fig. 11 Selected conglomerate-forming chiral molecules that racemise differently in solution. pic is 3-picoline. | ||
trans-Succinimides undergo racemisation through the achiral cis-isomers which possess an intrinsic mirror plane (Fig. 11d).43 Starting with the achiral cis-isomer, desymmetrisation facilitated by DBU provides both enantiomers which rapidly racemise through the cis-isomer. In combination with conglomerate crystallisation, this resulted in total spontaneous resolution to give the products in quantitative yields and 85–98% ee.
In addition to chiral molecules, achiral molecules were recently obtained in enantiopure form through Viedma ripening. Of all achiral compounds, between 8 and 13% crystallise as conglomerate crystals. McLaughlin et al. applied Viedma ripening to deracemise ten achiral organic molecules that crystallise in a chiral fashion (three examples are shown in Fig. 12).44 Solid-state circular dichroism (CD) spectroscopy was used to determine the solid state ee.
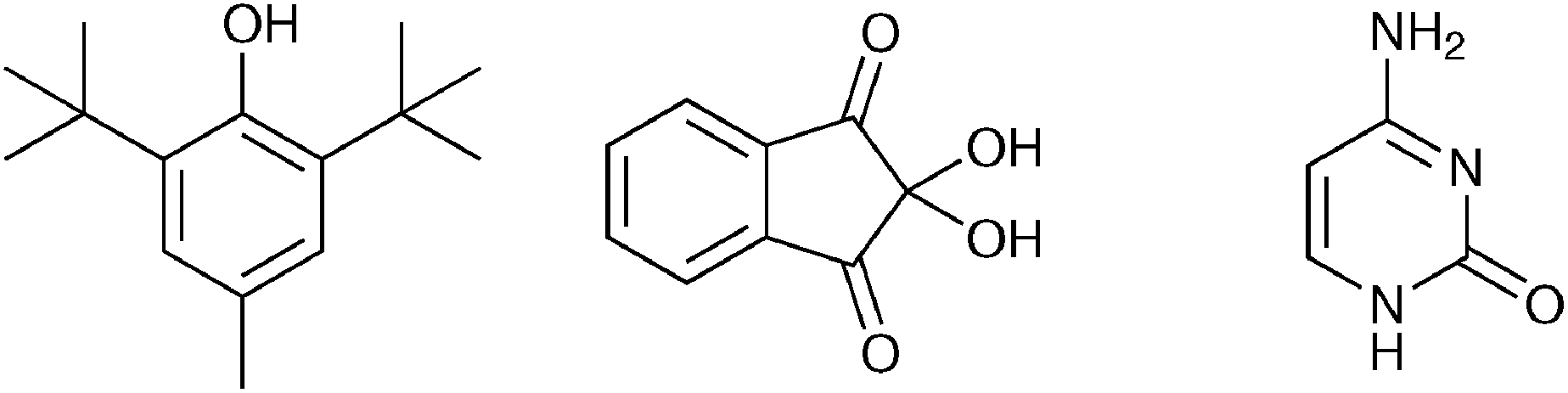 | ||
| Fig. 12 Selected achiral molecules which form conglomerate crystals. | ||
Besides racemisation in solution, the formation of racemic conglomerate crystals is another prerequisite for reaching single chirality using the interplay between crystals and a solution. The synthesis of a library of derivatives is one approach to find conglomerate crystals. On the other hand, tailoring crystallisation processes can also open up possibilities that could enable the formation of additional examples of conglomerate crystals. This way, Spix et al. showed that a metastable conglomerate of glutamic acid can be deracemised through Viedma ripening (Fig. 13).45 Although glutamic acid forms a racemic compound in the form of a hydrate, it was found that a metastable conglomerate form can be obtained as the kinetic product in the presence of acetic acid. Racemisation of glutamic acid proceeds at elevated temperatures which also facilitates formation of two other unwanted racemic forms of the product. Therefore, experimental conditions were tweaked which ultimately enabled access to enantiopure glutamic acid in 80% yield through Viedma ripening starting from low initial ee's of 14%.
 | ||
| Fig. 13 Deracemisation of a metastable conglomerate through Viedma ripening. | ||
Alternatively, salt formation can be used as a tool to prepare a library of compounds which in turn could provide new conglomerate candidates. This way, Spix et al. combined six different amino acids with four different sulfonates resulting in twenty-four different salts.46 It was found that three out of the twenty-four salts (13%) were racemic conglomerate crystals. Although leucine-2,5-xylenesulfonate was unstable under the applied Viedma ripening conditions which involves the use of acetic acid, the two other conglomerates (i.e. alanine-4-chlorobenzenesulfonate in 35–42% yield and phenylalanine-2,5-xylenesulfonate in 60–63% yield) could be deracemised through Viedma ripening (Fig. 14). In all these cases, the yield is somewhat limited due to the solubility of the compounds.
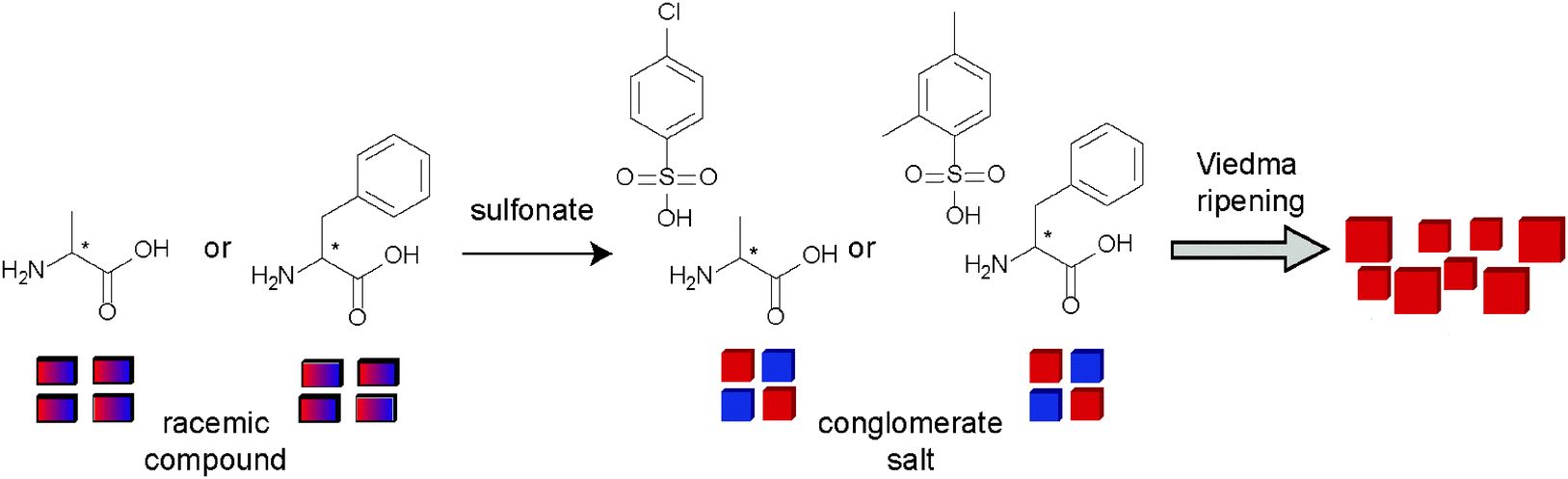 | ||
| Fig. 14 Amino acid salts that can be deracemised through Viedma ripening. | ||
While Viedma ripening found broad applicability, Viedma himself in collaboration with other groups found that deracemisation can be achieved through the use of a temperature gradient at high temperatures and without grinding.47,48 Coquerel et al. extended this observation to the use of deliberate ‘temperature cycling’ in order to gain a clearer picture of the mechanism behind this form of deracemisation, “of which the mechanism is still matter of debate”.49,50 With temperature cycling, all crystals start to dissolve simultaneously during a heating period, while the crystals remaining after the heating cycle are subsequently grown during the cooling period by consuming the excess of the solute molecules in the supersaturated solution (Fig. 15). A typical temperature program for temperature cycling involves rapid heating but slow cooling to avoid primary nucleation of the unwanted enantiomer. Such a program is repeated several times until an ee of 100% is reached.
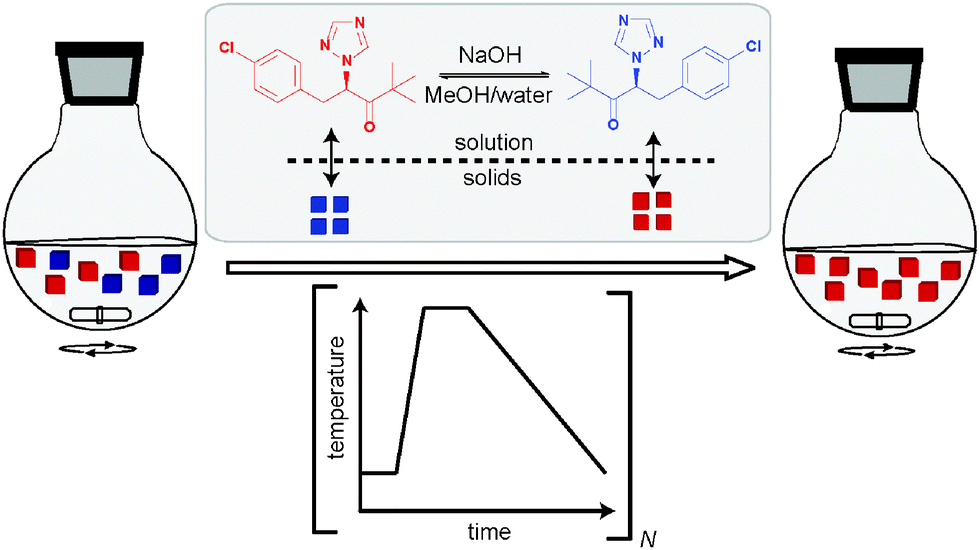 | ||
| Fig. 15 Deracemisation through repeated heating and cooling of a suspension. | ||
(5) Conclusions and outlook
In this tutorial review we have shown that molecules that form conglomerate crystals and racemise in solution can easily be obtained in enantiopure form through total spontaneous resolution, Viedma ripening or variants thereof. The main bottleneck that prevents a wider application of these techniques remains the prerequisite of conglomerate crystallisation. Currently, it is unknown why most compounds crystallise as racemic compounds instead of racemic conglomerate crystals. Therefore, the most straightforward method to acquire conglomerates is to prepare a library of derivatives through synthesis or salt formation. Libraries of crystals could also be prepared through formation of co-crystals or solvates. Provided that the library is sufficiently large, a number of derivatives should turn out to be conglomerate crystals. The second prerequisite is racemisation in solution, though this is not needed for achiral molecules that crystallise as a racemic conglomerate. Recent reports have shown that various racemisation approaches can be applied in combination with Viedma ripening and total spontaneous resolution, leading to even more examples of enantiopure products.Even though there still is no absolute consensus, recent mechanistic studies have provided a deeper understanding of the driving forces behind Viedma ripening. Most studies have shown that a feedback mechanism must be involved to account for the exponential increase in ee. This feedback mechanism can be explained in terms of chiral clusters which selectively incorporate into crystals of the same handedness. Despite some indirect experimental observations, undisputed evidence for chiral clusters remains to be revealed.
After the discovery of the traditional Viedma ripening method, many novel applications have been developed which enable the use of Viedma ripening on a large industrial scale as well as in synthetic organic chemistry. Also, attrition is no longer required as gentle temperature fluctuations can lead to solid-state deracemisation. Crystal-solution systems are also very useful in asymmetric autocatalysis as enantiopure compounds can be obtained from achiral reactants.
From this review it appears that to date many compounds are still obtained in enantiopure form through total spontaneous resolution instead of Viedma ripening. However, it should be noted that Viedma ripening is a more robust and reliable method as crystals of the unwanted enantiomer, which often prevent total spontaneous resolution from reaching an enantiopure end state, are transformed into the desired enantiomer. Although seeding is used in total spontaneous resolution to obtain the best results in terms of chiral purity of the final product, Viedma ripening leads to complete deracemisation without seeding, even when starting from a racemic mixture of crystals.
Notes and references
- I. Myrgorodska, C. Meinert, Z. Martins, L. Le Sergeant d'Hendecourt and U. J. Meierhenrich, Angew. Chem., Int. Ed., 2015, 54, 1402–1412 CrossRef CAS PubMed
.
- V. Balzani, A. d. Meijere, K. N. Houk, H. Kessler, J. M. Lehn, S. V. Ley, S. L. Schreiber, J. Thiem, B. M. Trost, F. Vögtle and H. Yamamoto, Top. Curr. Chem., 2007, 269, 1–313 CrossRef
- W. L. Noorduin, E. Vlieg, R. M. Kellogg and B. Kaptein, Angew. Chem., Int. Ed., 2009, 48, 9600–9606 CrossRef CAS PubMed
- T. Buhse, D. Durand, D. Kondepudi, J. Laudadio and S. Spilker, Phys. Rev. Lett., 2000, 84, 4405–4408 CrossRef CAS
- G. Coquerel, Chem. Soc. Rev., 2014, 43, 2286–2300 RSC
-
E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, John Wiley & Sons, Inc., 1994 Search PubMed
- L. C. R. Pasteur, Hebd. Séanc. Acad. Sci. Paris, 1848, 535 Search PubMed
- J. E. Hein, B. Huynh Cao, C. Viedma, R. M. Kellogg and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134, 12629–12636 CrossRef CAS PubMed
- E. Havinga, Chem. Weekbl., 1941, 38, 642–644 CAS
- E. Havinga, Biochim. Biophys. Acta, 1954, 13, 171–174 CrossRef CAS
- F. C. Frank, Biochim. Biophys. Acta, 1953, 11, 459–463 CrossRef CAS
- D. K. Kondepudi, R. J. Kaufman and N. Singh, Science, 1990, 250, 975–976 CAS
- R. E. Pincock, R. R. Perkins, A. S. Ma and K. R. Wilson, Science, 1971, 174, 1018–1020 CAS
- D. K. Kondepudi, K. L. Bullock, J. A. Digits, J. K. Hall and J. M. Miller, J. Am. Chem. Soc., 1993, 115, 10211–10216 CrossRef CAS
- D. J. Durand, D. K. Kondepudi, P. F. Moreira Jr and F. H. Quina, Chirality, 2002, 14, 284–287 CrossRef CAS PubMed
- D. K. Kondepudi and K. Asakura, Acc. Chem. Res., 2001, 34, 946–954 CrossRef CAS PubMed
- C. Viedma, J. Cryst. Growth, 2004, 261, 118–121 CrossRef CAS PubMed
- C. Viedma, Phys. Rev. Lett., 2005, 94, 065504 CrossRef
- W. L. Noorduin, T. Izumi, A. Millemaggi, M. Leeman, H. Meekes, W. J. P. Van Enckevort, R. M. Kellogg, B. Kaptein, E. Vlieg and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 1158–1159 CrossRef CAS PubMed
- W. L. Noorduin, P. van der Asdonk, A. A. C. Bode, H. Meekes, W. J. P. van Enckevort, E. Vlieg, B. Kaptein, M. W. van der Meijden, R. M. Kellogg and G. Deroover, Org. Process Res. Dev., 2010, 14, 908–911 CrossRef CAS
- W. L. Noorduin, B. Kaptein, H. Meekes, W. J. P. van Enckevort, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2009, 48, 4581–4583 CrossRef CAS PubMed
- M. W. van der Meijden, M. Leeman, E. Gelens, W. L. Noorduin, H. Meekes, W. J. P. van Enckevort, B. Kaptein, E. Vlieg and R. M. Kellogg, Org. Process Res. Dev., 2009, 13, 1195–1198 CrossRef CAS
- B. Kaptein, W. L. Noorduin, H. Meekes, W. J. P. van Enckevort, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2008, 47, 7226–7229 CrossRef CAS PubMed
- W. L. Noorduin, H. Meekes, W. J. P. van Enckevort, B. Kaptein, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2010, 49, 2539–2541 CrossRef CAS PubMed
- W. L. Noorduin, W. J. P. van Enckevort, H. Meekes, B. Kaptein, R. M. Kellogg, J. C. Tully, J. M. McBride and E. Vlieg, Angew. Chem., Int. Ed., 2010, 49, 8435–8438 CrossRef CAS PubMed
- M. Uwaha, J. Phys. Soc. Jpn., 2004, 73, 2601–2603 CrossRef CAS
- C. Viedma, J. M. McBride, B. Kahr and P. Cintas, Angew. Chem., Int. Ed., 2013, 52, 10545–10548 CrossRef CAS PubMed
- Z. El-Hachemi, J. Crusats, J. M. Ribo, J. M. McBride and S. Veintemillas-Verdaguer, Angew. Chem., Int. Ed., 2011, 50, 2359–2363 CrossRef CAS PubMed
- W. L. Noorduin, H. Meekes, W. J. P. van Enckevort, A. Millemaggi, M. Leeman, B. Kaptein, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2008, 47, 6445–6447 CrossRef CAS PubMed
- W. L. Noorduin, A. A. C. Bode, M. van der Meijden, H. Meekes, A. F. van Etteger, W. J. P. van Enckevort, P. C. M. Christianen, B. Kaptein, R. M. Kellogg, T. Rasing and E. Vlieg, Nat. Chem., 2009, 1, 729–732 CrossRef CAS PubMed
- K. Asakura, K. Kobayashi, Y. Mizusawa, T. Ozawa, S. Osanai and S. Yoshikawa, Phys. D, 1995, 84, 72–78 CrossRef CAS
- K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767–768 CrossRef CAS PubMed
- K. Soai, I. Sato, T. Shibata, S. Komiya, M. Hayashi, Y. Matsueda, H. Imamura, T. Hayase, H. Morioka, H. Tabira, J. Yamamoto and Y. Kowata, Tetrahedron: Asymmetry, 2003, 14, 185–188 CrossRef CAS
- J. B. Hendrickson, J. Am. Chem. Soc., 1975, 97, 5784–5800 CrossRef CAS
- R. R. E. Steendam, J. M. M. Verkade, T. J. B. van Benthem, H. Meekes, W. J. P. van Enckevort, J. Raap, F. P. J. T. Rutjes and E. Vlieg, Nat. Commun., 2014, 5, 6543 Search PubMed
- S. B. Tsogoeva, S. Wei, M. Freund and M. Mauksch, Angew. Chem., Int. Ed., 2009, 48, 590–594 CrossRef CAS PubMed
- A. M. Flock, C. M. M. Reucher and C. Bolm, Chem. – Eur. J., 2010, 16, 3918–3921 CrossRef CAS PubMed
- A. Lennartson, S. Olsson, J. Sundberg and M. Håkansson, Angew. Chem., Int. Ed., 2009, 48, 3137–3140 CrossRef CAS PubMed
- A. Lennartson and M. Håkansson, Angew. Chem., Int. Ed., 2009, 48, 5869–5871 CrossRef CAS PubMed
- M. Sakamoto, F. Yagishita, M. Ando, Y. Sasahara, N. Kamataki, M. Ohta, T. Mino, Y. Kasashima and T. Fujita, Org. Biomol. Chem., 2010, 8, 5418–5422 CAS
- F. Yagishita, H. Ishikawa, T. Onuki, S. Hachiya, T. Mino and M. Sakamoto, Angew. Chem., Int. Ed., 2012, 51, 13023–13025 CrossRef CAS PubMed
- R. R. E. Steendam, M. C. T. Brouwer, E. M. E. Huijs, M. W. Kulka, H. Meekes, W. J. P. van Enckevort, J. Raap, F. P. J. T. Rutjes and E. Vlieg, Chem. – Eur. J., 2014, 20, 13527–13530 CrossRef CAS PubMed
- S. Hachiya, Y. Kasashima, F. Yagishita, T. Mino, H. Masu and M. Sakamoto, Chem. Commun., 2013, 49, 4776–4778 RSC
- D. T. McLaughlin, T. P. T. Nguyen, L. Mengnjo, C. Bian, Y. H. Leung, E. Goodfellow, P. Ramrup, S. Woo and L. A. Cuccia, Cryst. Growth Des., 2014, 14, 1067–1076 CAS
- L. Spix, H. Meekes, R. H. Blaauw, W. J. P. van Enckevort and E. Vlieg, Cryst. Growth Des., 2012, 12, 5796–5799 Search PubMed
- L. Spix, A. Alfring, H. Meekes, W. J. P. van Enckevort and E. Vlieg, Cryst. Growth Des., 2014, 14, 1744–1748 CAS
- C. Viedma and P. Cintas, Chem. Commun., 2011, 47, 12786–12788 RSC
- C. Viedma, J. E. Ortiz, T. de Torres, T. Izumi and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 15274–15275 CrossRef CAS PubMed
- K. Suwannasang, G. Coquerel, C. Rougeot and A. E. Flood, Chem. Eng. Technol., 2014, 37, 1329–1339 CrossRef CAS PubMed
- K. Suwannasang, A. E. Flood, C. Rougeot and G. Coquerel, Cryst. Growth Des., 2013, 13, 3498–3504 CAS
| This journal is © The Royal Society of Chemistry 2015 |