
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhysiological concentrations of albumin favor drug binding†
D.
Tatlidil
a,
M.
Ucuncu
b and
Y.
Akdogan
*a
aMaterials Science and Engineering Department, Ízmir Institute of Technology, 35430 Urla, Ízmir, Turkey. E-mail: yasarakdogan@iyte.edu.tr
bDepartment of Chemistry, Ízmir Institute of Technology, 35430 Urla, Ízmir, Turkey
First published on 29th July 2015
Abstract
The ability to track drug binding and release makes electron paramagnetic resonance (EPR) spectroscopy well suited for drug delivery studies. Using the continuous wave (cw) EPR technique to extract information about the dynamics of the spin labeled drugs we can simultaneously determine the bound and unbound drugs. Here, spin labeled salicylic acid (SLSA) binding to and release from bovine serum albumin (BSA) is investigated, as a model for drug–transport protein interaction. We studied SLSA–BSA binding in a wide concentration range and found that the stoichiometry of the drug–protein increases significantly when the physiological range of BSA concentration is reached. Our EPR results explicitly reveal that up to ∼7 SLSA can bind to one albumin at the physiological concentration, whereas at lower BSA concentrations (<0.125 mM) the SLSA–BSA stoichiometry is maximum 2. Moreover, we studied drug release and showed that the ratio of bound to unbound SLSA concentrations remains relatively stable during dialysis. This indicates that the binding equilibrium of SLSA is not altered through the process of dialysis. This study demonstrates that cw EPR spectroscopy in combination with spin labeled drugs is an effective technique for binding and release studies and stoichiometric analysis of drug–protein interactions.
Introduction
The extent of drug–transport protein binding is important to determine the drug dose. Drugs can travel in the system either as free floating molecules or as protein bound molecules in a reversible manner. Mostly, the unbound drug concentration, rather than the protein bound concentration, is used to determine the effect of a drug.1,2 On the other hand, protein binding prevents drugs to be metabolized completely or to reach high toxic levels. The bound fraction is released slowly to maintain equilibrium. Binding and release of drugs are altered in a range of physical and chemical conditions.3,4 Therefore, understanding the factors that affect the drug binding to and release from proteins in the plasma or tissue is very important.Several spectroscopic techniques have been used in order to study the interaction between drugs and proteins, including fluorescence,5 UV-vis,6 FTIR6 and NMR7 spectroscopies. Some of these techniques have been used only to observe the changes in proteins instead of drugs in the protein-drug complexes. For example, the intrinsic fluorescence of bovine serum albumin (BSA) was monitored by fluorescence emission spectroscopy in the presence of aspirin.5 Since aspirin binding or interaction quenches the fluorescence of albumin, fluorescence spectroscopy is used as an indirect method to observe the binding of aspirin to albumin. Similarly, interactions of vitamin C and aspirin with BSA were studied by FTIR to examine the variations in the stability and conformation of BSA.6 In the same study, UV-vis spectroscopy was used to calculate the binding constants of the drugs by considering the absorbance values of drugs, BSA and drug–BSA complexes.6 Since absorption wavelengths of free aspirin, free BSA and the aspirin–BSA complex are very similar, it is difficult to distinguish the bound/unbound ratio of aspirin in the final aspirin–BSA complex using UV-vis spectroscopy. Differently, in the NMR spectroscopy, changes in the chemical shifts and relaxation rates of a particular part of drugs can be the proof of a drug–protein complex formation.7 The part of the drug which strongly binds to protein has longer correlation time than the other parts of the drug. Therefore, NMR allows direct monitoring of the site of interest in the drug. However, such studies cannot simultaneously display the signatures of the bound and unbound drug molecules, especially within the physiological range of albumin.
Electron paramagnetic resonance (EPR) spectroscopy in combination with spin labeling is a powerful method for studying the interaction between small drugs and biomacromolecules.9,10 Since most of the drugs are diamagnetic and contain no paramagnetic centres, exogenous stable free radicals called spin labels are used as reporters. Tempo (2,2,6,6-tetramethylpiperidine-1-oxyl) based nitroxide radicals, the most commonly employed spin label, can be attached to a proper functional group on the drug (Fig. 1). Spin labeled drugs have been synthesized and used to study the drug mechanisms at a molecular level for a long time.11–13 Besides their reporter properties, nitroxides are also used in anti-oxidant therapies for several diseases because they can effectively scavenge the reactive oxygen species and therefore improve cell viability.14,15
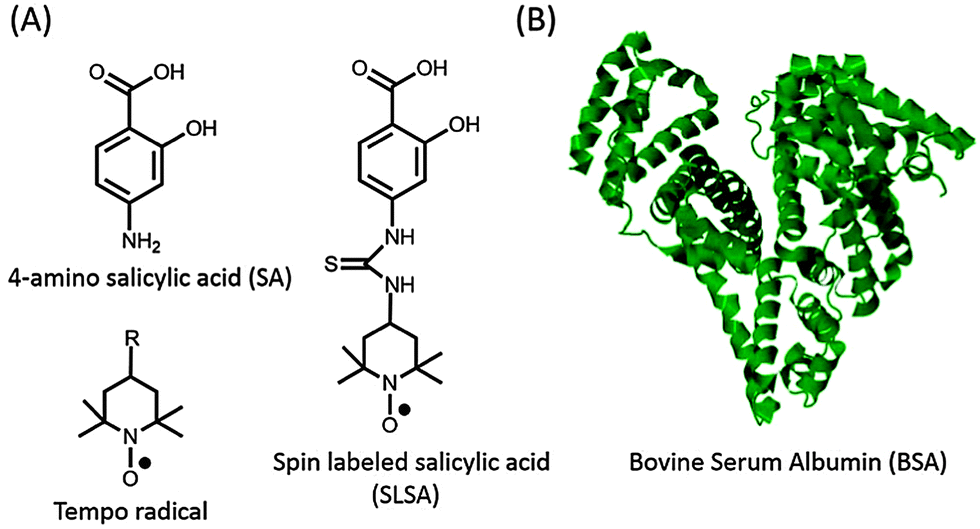 | ||
| Fig. 1 (A) Chemical structures of 4-amino salicylic acid (SA), Tempo based nitroxide radicals (R: –OH, –NH2, –COOH, –O, –NCS, etc.) and spin labeled salicylic acid (SLSA). (B) Crystal structure (pdb-ID: 3v03) of bovine serum albumin (BSA).8 | ||
Here, we use continuous wave (cw) EPR spectroscopy as an alternative technique to study the interaction between drugs and proteins. One major advantage of cw EPR spectroscopy is the ability to observe the unbound drug and the bound drug simultaneously, due to its sensitivity to the rotational dynamics of the radicals.11–13 Bound spin labeled molecules (immobilized) have characteristic broad signals stemming from restricted rotational motion, and unbound spin labeled molecules (mobilized) have sharp three-line signals coming from freely tumbling motion.16–18
Salicylic acid (SA), which is a non-steroidal anti-inflammatory drug, and serum albumin, which is the most abundant plasma protein, were used as a model drug and transporter protein, respectively. SA contains two functional groups, a carboxylic acid (–COOH) group and a hydroxyl (–OH) group (Fig. 1). In order to preserve these functional groups, spin label is covalently attached to the phenyl group of SA, and so that spin labeled salicylic acid (SLSA) is synthesized (Fig. 1 and Fig. S1, ESI†). Human serum albumin (HSA) with three α-helical domains (I–III) serves as a carrier of many drugs from the bloodstream to target cells.19 The domains II and III contain two principal ligand binding sites, known as Sudlow's sites I and II, respectively.20 Furthermore, several other binding sites were also observed including seven long fatty acid binding sites.21,22 Bovine serum albumin (BSA) has been widely used in experiments due to its structural homology to HSA (76% of amino acid sequence homology) and its lower cost (Fig. 1).5–7
Here, we specifically aim at studying in vitro SLSA binding and release profiles of BSA as well as the SLSA capacity of BSA at different albumin concentrations. Dynamics of drugs changes enormously upon protein binding so we can monitor the bound and unbound spin labeled drug in the presence of albumin simultaneously by EPR spectroscopy.
Experimental section
Reagents and instruments
Bovine serum albumin (fatty acid free), 4-amino salicylic acid, Tempo-4-amino, hydrochloric acid (37%, 12.2 M), dichloromethane (99.8%), trimethylamine (99.5%), ethyl acetate (99.5%), methanol (99.8%) were purchased from Sigma-Aldrich. Thiophosgene (99%) was purchased from Merck. All reagents were used without further purification. All solvents were of analytical-reagent grade. Doubly distilled water was used throughout the experiments.The samples were measured on a Varian Cary Eclipse Fluorescence spectrophotometer equipped with 1.0 cm path length quartz cuvettes and a CMS 8400 (Adani) benchtop X-band EPR spectrometer with a TE102 resonator cavity.
Synthesis of spin labeled salicylic acid (SLSA)
4-Amino salicylic acid (0.01 mol) was dissolved in 32 mL water and then acidified with 3.4 mL HCl (12.2 M) in a 100 mL reaction tube with a stopcock. Thereafter, thiophosgene (0.044 mol) was introduced. After stirring for 1 hour at 0 °C (in an ice bath), the solution was stirred for 2.5 hours at 25 °C. The solution was filtered and dried under vacuum. 4-Isothiocyanate salicylic acid was isolated as a white powder. The yield of the experiment was 50% (Fig. S1, ESI†).Tempo-4-amino (0.625 mmol) and 4-isothiocyanate salicylic acid (0.46 mmol) were dissolved in 20 mL DCM in a 50 mL flask. A few drops of triethylamine were added and stirred overnight at 25 °C. Tempo attached salicylic acid (spin labeled salicylic acid, SLSA) was purified with column chromatography (ethyl acetate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol, 15:1, 13:1, 12:1 and 10:1). The yield of the experiment was 46% (Fig. S1, ESI†).23
:methanol, 15:1, 13:1, 12:1 and 10:1). The yield of the experiment was 46% (Fig. S1, ESI†).23
Binding of SLSA to BSA
Aqueous solutions of BSA at different concentrations were prepared in 0.1 M phosphate buffer (pH 7.4) in 10 mL vials. The SLSA stock solution (1 M) was prepared in DMSO in a 0.2 mL pcr tube. The desired ratio of SLSA to BSA was obtained with the addition of an appropriate amount of SLSA solution to the protein solution with constant stirring to ensure the formation of a homogeneous solution. In the final solution, the DMSO concentration was kept below 1% (v/v). The solution was allowed to equilibrate at 37 °C for 20 min for the fluorescence and EPR measurements.Release of SLSA from BSA
SLSA release from BSA was studied in phosphate buffer (0.1 M, pH 7.4) at 25 °C and 37 °C. 3 mL, 0.5 mM SLSA in BSA solution (SLSA/BSA, 1:1) was placed in a D-tube dialyzer (Merck, MWCO 6–8 kDa), and then the solution was placed in a beaker containing 1000 mL release phosphate buffer (0.1 M, pH 7.4) under stirring at 1500 rpm. At prefixed times a 7 μL sample was removed from the D-tube dialyzer and measured by EPR spectroscopy to determine the amount of SLSA diffused as well as the ratio of bound/unbound SLSA in the albumin solution.
Fluorescence measurements
Fluorescence measurements were carried out using a Varian Cary Eclipse Fluorescence spectrophotometer equipped with 1.0 cm path length quartz cuvettes. Upon excitation at 278 nm, the emission spectra were collected in the range of 300–550 nm. The slit width was 5 nm for both excitation and emission. 10 μL salicylic acid (SA) or spin labeled salicylic acid (SLSA) (6 × 10−2 mM) was added to 3.0 mL BSA (2 × 10−4 mM) in phosphate buffer. For the bare BSA measurement, 10 μL buffered solution was also added to 3.0 mL BSA. Similarly, bare SA and SLSA were measured in the absence of BSA.EPR measurements
A CMS 8400 (Adani) benchtop spectrometer provided with a TE102 resonator cavity was used for all X-band EPR measurements at a microwave frequency of ∼9.4 GHz. Experiments were performed at 25 °C using a modulation amplitude of 0.1 mT. Measurements were performed in quartz capillary sample tubes.The shown EPR spectra were normalized to the intensity of the high field line of the unbound SLSA. In the EPR spectrum, the high field line of the unbound SLSA is the least affected signal by the presence of bound SLSA's signals. All spectra were simulated with the Matlab-based Easyspin 4.5.5 software package.24
Results and discussion
We have synthesized the nitroxide derivative of the SA to study the binding of the SA drug to BSA protein. In the synthesis of SLSA, we used 4-amino SA to preserve the functional groups of SA (–COOH and –OH) (Fig. S1, ESI†). First, 4-amino SA was transformed into 4-isothiocyanate SA. In the following step, the nitroxide based Tempo radical was attached to the isothiocyanate group of SA. Tempo radicals have excellent stability at both physiological temperature and pH, and are commercially available with several functional groups at the 4-position (Fig. 1).A crystallographic structural study of the HSA–SA complex showed that the carboxylate group of SA forms hydrogen bonds with surrounding amino acids in site I.25 Moreover, the coordination of hydroxyl and phenyl groups of SA with hydrophobic residues of HSA was also reported.25 Here, spin labeling of the SA increases the hydrophobicity of the SA while keeping the carboxylate and hydroxyl groups still active. Considering these possible interactions between SA and BSA, it is not surprising that the more hydrophobic nitroxide derivative of the SA (SLSA) could bind to BSA.
Fluorescence spectroscopy studies on the binding of SA and SLSA to BSA
In order to compare the binding ability of SA and SLSA to BSA, fluorescence emission spectra of BSA were recorded in the absence and presence of SA and SLSA (Fig. 2). | ||
| Fig. 2 Fluorescence emission spectra of BSA (black) in the presence of SA (red) and SLSA (blue). The concentration of BSA is 2 × 10−7 M in the 0.1 M phosphate buffer at a drug/BSA ratio of 1. The dashed lines belong to the fluorescence emission of SA (red) and SLSA (blue) in the buffer solutions without BSA. | ||
Intrinsic tryptophan fluorescences (Trp-212 and Trp-134) dominate the BSA fluorescence spectrum with a maximum at 350 nm. Moreover, SA has a characteristic fluorescence emission spectrum with a maximum at around 410 nm. However, fluorescence emission intensity of SLSA is about 11 times weaker than that of SA because spin labeling of SA quenched the fluorescent signal of the SA due to the interaction between the nitroxide radical and the fluorophore (Fig. 2, dashed lines).
In general, the fluorescence intensity of BSA decreases when other molecules bind to BSA due to the distortion of the microenvironment around the tryptophan residues of BSA.5,26 Ni et al. showed that the addition of SA to BSA decreases the fluorescence intensity of BSA at 350 nm, and a second peak appears at ∼410 nm.5Fig. 2 shows that the fluorescence intensity of BSA (2 × 10−4 mM) decreases upon addition of SA and SLSA at a drug/BSA ratio of 1. However, the second peak at 410 nm appears only upon addition of SA. Unambiguously, the decreasing ratios in fluorescence intensity of BSA are very similar after SA and SLSA contributions. This observation suggested that BSA binding abilities of SA and SLSA are very comparable.
Furthermore, Fig. S2 (ESI†) showed the fluorescence emission spectra of BSA (1.5 × 10−7 M) in the presence of SA with different concentrations (0–4.3 × 10−7 M) at 303 K. The fluorescence band intensity of BSA decreases (quenches) upon addition of SA. At the same time, a second peak corresponding to SA appears gradually.
The quenching is generally characterized by the Stern–Volmer equation.27
| F0/F = 1 + KSV[Q] | (1) |
:[BSA] is 1.5) and the quenching ceased after 3.2 × 10−7 M SA concentration which is the limit of detection (the ratio of [SA]:[BSA] is 2.4) (Fig. S2, ESI†).
EPR spectroscopy studies on the binding of SLSA to BSA
In order to study SLSA–BSA binding in detail, first we characterized the EPR spectrum of SLSA. X-band cw EPR spectra of 0.6 mM SLSA and Tempo-4-amino in 0.1 M phosphate buffer at 25 °C are presented in Fig. S3 (ESI†). The usual splitting in three hyperfine lines comes from the nitrogen nucleus with nuclear spin I = 1 of the nitroxide group. The EPR parameters of SLSA and Tempo-4-amino (isotropic g-values (giso), isotropic hyperfine coupling constants (Aiso) and rotational correlation times (τR)) were derived from their spectral simulations. Both have similar g-values and hyperfine values (giso: 2.00554 vs. 2.00552 and Aiso: 1.70 mT vs. 1.68 mT, for SLSA vs. Tempo-4-amino, respectively).The line shape of an EPR spectrum contains information about the rotational dynamics of a paramagnetic centre. For both samples, the low-field and central lines of spectra are almost equal, but the high field line is less intense for the SLSA. This indicates the slower rotational motion of SLSA (τR = 0.07) compared to the rotational motion of Tempo-4-amino (τR = 0.02 ns) (Fig. S3, ESI†). This suggests that binding of SA to Tempo-4-amino restricts the motion of the radical centre.
Next, we studied SLSA binding to BSA using cw EPR spectroscopy (Fig. 3 and 4). In solution, the nitroxide line shape of the cw EPR spectrum of the spin labeled drug is strongly influenced by the protein binding. Rotational freedom of the radical center on the molecule significantly decreases upon macromolecule binding so that the rotational correlation time increases from the ps to μs range as a result of the immobilization strength.28 In a typical room temperature EPR spectrum, the unbound (mobilized) spin labeled drug possesses sharp three-line signals, signatures of freely tumbling motion. In contrast, the protein bound (immobilized) spin labeled drug gives signals with broad outer hyperfine features stemming from restricted rotational motion (Fig. 3). Therefore, the EPR spectroscopy technique allows simultaneous detection of bound and unbound spin labeled drug molecules from their corresponding signals.
 | ||
| Fig. 3 (1) Cw EPR spectrum of 0.6 mM SLSA in BSA solution (1:1) (black) and its simulation (red). The simulation of the spectrum was obtained by the addition of simulated unbound SLSA (2) and simulated bound SLSA (3). | ||
 | ||
| Fig. 4 (A) Superposition of simulations of unbound (black) and bound (red) fractions of the EPR spectrum belongs to 0.6 mM SLSA in BSA solution (1:1). Single integrations (B) and double integrations (C) of EPR spectra in (A). Double integration is performed to determine the proportion of the bound and unbound SLSA to the total. | ||
Moreover, the ratio of bound to unbound spin labeled drugs, and the number of bound SLSA molecules per albumin molecule can be calculated from the areas under the EPR signals (obtained by double integrations of EPR spectra, Fig. 4):
 | (2) |
Fig. 3 shows the cw EPR spectrum of 0.6 mM SLSA in the BSA solution at a SLSA/BSA ratio of 1 with the corresponding simulation. The simulation of the spectrum was obtained by the addition of the simulated three line spectrum (unbound SLSA, τR = 0.07 ns) and simulated broad signals (bound SLSA, τR = 10 ns). Double integrations of the simulated EPR spectra of unbound and bound SLSA revealed that ∼80% of SLSA is bound to albumin (Fig. 4). As a control experiment, a mixture of Tempo-4-amino and BSA solution was measured and only three sharp signals coming from free Tempo-4-amino radicals were obtained (Fig. S4, ESI†). This indicates that Tempo-4-amino does not bind to BSA by itself.
Concentration effects on the protein binding
In solution, the extent of drug–protein conjugation increases when the concentrations of drug and protein increase according to the collision theory.29Fig. 5A shows the cw EPR spectrum of a concentrated drug–protein solution with 3.0 mM SLSA in 3.0mM BSA solution (1:1) (red line). Most of the SLSA are bound to BSA with a fraction of bound SLSA as 0.95 (Fig. 5B). When the SLSA–BSA solution is gradually diluted with buffer while keeping the SLSA/BSA ratio at unity, the fraction of bound SLSA decreases from 0.95 to 0.50 at 3.0 mM and 0.1 mM BSA, respectively. This suggests that dilution causes the release of SLSA from BSA.
 | ||
| Fig. 5 (A) Cw EPR spectrum of SLSA in BSA solution at a SLSA/BSA ratio of 1. The concentrations of SLSA and BSA from top to bottom are 3.0, 2.5, 2.0, 1.5, 1.0, 0.5 and 0.25 mM, and the free 0.6 mM SLSA in buffer without BSA (gray color). The EPR spectra are normalized to the intensity of the high field line of the unbound SLSA. (B) Fraction of bound SLSA obtained from simulated spectra of 3.0, 2.75, 2.5, 2.0, 1.5, 1.0, 0.85, 0.7, 0.6, 0.5, 0.4, 0.3, 0.25, 0.18 and 0.10 mM SLSA in BSA solutions at a SLSA/BSA ratio of 1. The circle shows the concentrations of BSA in the physiological range (0.5–0.7 mM BSA). | ||
Usually, drug binding is a reversible process in which drugs bind to the albumin with weak chemical bonds, such as hydrogen bonds or van der Waals forces.25,30 We have also an equilibrium between bound and unbound SLSA in the BSA solution. During the process of diluting the system to lower concentrations, the collision probability of unbound SLSA and BSA decreases which reduces the formation of SLSA–BSA conjugation. In order to maintain the equilibrium between bound and unbound drugs, BSA releases more SLSA. Thus, the fraction of unbound SLSA increases and the fraction of bound SLSA decreases regularly. When we plotted the fraction of bound SLSA in 1:1 SLSA:BSA solutions of varying BSA concentrations (0.1 mM–3.0 mM) we obtained a saturation curve. The bound fraction is saturated just above the physiological concentrations of albumin (0.5–0.7 mM). However, below the physiological concentrations of albumin the fraction of bound SLSA decreases promptly.
The experimental results show that SLSA can bind to BSA with a very high efficiency at the physiological concentrations of BSA. But, below the physiological concentrations, BSA loses its affinity for SLSA dramatically.
We measured the EPR spectra of 2 mM, 0.8 mM and 0.4 mM of SLSA in BSA solution at a SLSA/BSA ratio of 1 within a period of time after preparation of the samples from 1 minute to 30 minutes (Fig. S5, ESI†). Since tuning the EPR cavity after sample insertion and EPR measuring collectively take some time, we cannot collect the data immediately (0 min).
Fig. S5 (ESI†) shows that EPR spectra of different concentrations of SLSA/BSA (1:1) assemblies do not change with time. The bound to free drug ratio is constant within 30 min. Usually, drug–protein binding interaction is a kinetically rapid reversible interaction. Therefore, the quick drug association and dissociation rates in ranges of milliseconds and seconds could not be calculated by the EPR technique.
Binding stoichiometry of SLSA to BSA
Simulations of EPR spectra of both bound and unbound SLSA were used to obtain a maximum number of bound SLSA to BSA, at physiological albumin concentration. For this purpose, SLSA was added to 0.5 mM BSA solution to generate a series of samples with different SLSA concentrations from 0.5 to 9.5 mM, and recorded using EPR spectroscopy.In the normalized EPR spectra, the intensity of bound SLSA signals decreases with increasing SLSA concentration (Fig. 6A). After simulations of each spectrum, the fractions of bound SLSA were found to be 0.8 and 0.4 in the presence of 0.5 mM and 9.5 mM SLSA, respectively (Fig. S6, ESI†).
 | ||
| Fig. 6 (A) Cw EPR spectra of SLSA in a constant 0.5 mM BSA solution at different SLSA/BSA ratios. The concentrations of SLSA are 0.5, 1.25, 2.5, 3.75, 5.0, 6.5, 8.0 and 9.5 mM (from top to bottom). 0.5 mM free SLSA in buffer solution is shown by grey color. The EPR spectra are normalized to the intensity of the high field line of the unbound SLSA. (B) Ratio of total SLSA concentration to BSA concentration is shown by black line, and the ratio of bound SLSA to BSA concentration is shown by red line. Bound SLSA concentrations are obtained from simulations of EPR spectra from (A). | ||
Although, increasing the concentration of SLSA decreases the bound fraction of SLSA (Fig. S6, ESI†), the number of bound SLSA per BSA increases (Fig. 6B). Nonetheless, a deviation from the straight line coming from the ratio of total SLSA to BSA concentrations is observed by increasing the concentration of SLSA. Eventually, SLSA forms a saturated complex with BSA at a molar ratio of ∼7:1 (SLSA:BSA) (Fig. 6B). This indicates that at physiological concentration (0.5–0.7 mM) BSA possesses about 7 binding sites for SLSA (eqn 2).
In another experiment, the concentration of BSA was changed while the concentration of SLSA was kept constant at 0.6 mM. EPR spectra of 0.6 mM SLSA in different concentrations of BSA solutions between 3.0–0.3 mM (Fig. 7A) and 0.24–0.02 mM (Fig. 7B) show that the bound fraction of SLSA decreases from 0.96 (at 3.0 mM BSA) to 0.08 (at 0.02 mM BSA) (Fig. S7, ESI†).
 | ||
| Fig. 7 Cw EPR spectra of constant 0.6 mM SLSA in different concentrations of BSA. The concentrations of BSA are 3.00, 2.40, 1.80, 1.20, 0.90, 0.60, 0.45 and 0.30 mM in (A) and 0.24, 0.12, 0.06, 0.03 and 0.02 mM in (B). 0.6 mM free SLSA in buffer solution was shown as a reference. The EPR spectra are normalized to the intensity of the high field line of the unbound SLSA. (C) Ratio of total SLSA concentration to BSA concentration is shown by black line and the ratio of protein bound SLSA concentration to BSA concentration is shown by red line. Protein bound SLSA ratio is obtained from simulation of EPR spectra from (A) and (B). | ||
The ratio of bound SLSA/BSA reaches a plateau when the BSA concentration is as low as 0. 125 mM. Therefore, at lower BSA concentrations (<0.125 mM) compared to the physiological range, the maximum binding ratio was found to be ∼2:1 for the SLSA:BSA interaction (Fig. 7C).
If we compare the bound fractions of SLSA in two BSA systems (in a physiological range and in a diluted system) at the same molar ratio of 19:1 (SLSA:BSA), the bound fractions are 0.40 and 0.12 for the SLSA in 0.5 mM BSA and in 0.03 mM BSA, respectively. Therefore, in the physiological range maximum ∼7 SLSA molecules bind per BSA, and in a diluted BSA system maximum ∼2 SLSA molecules bind per BSA even at a higher [SLSA]/[BSA] molar ratio of 30:1. Binding stoichiometries of small drugs to albumin obtained by fluorescence spectroscopy are also in line with our EPR results. Ni et al. reported the binding ratios of aspirin–BSA and ibuprofen–BSA as 3:1 and 2:1, respectively.5 Also, they studied the binding stoichiometry of SA to BSA and showed that SA primarily binds to site I and may have two additional low affinity binding sites in site I and II.31 One should be careful when comparing the results of EPR and fluorescence experiments. Since the fluorescence measurements were done in the presence of around 0.0002 mM BSA, we can compare those results with our EPR results obtained from the diluted BSA system (0.02 mM) not the physiological range (0.5–0.7 mM).
We have schematically summarized our EPR results in Fig. 8. A combination of SLSA–BSA dilution experiments (Fig. 5) and stoichiometry analyses (Fig. 6 and 7) shows that albumin can bind to maximum 7 SLSA at the physiological range. However, albumin loses its affinity for SLSA when the concentration of albumin decreases.
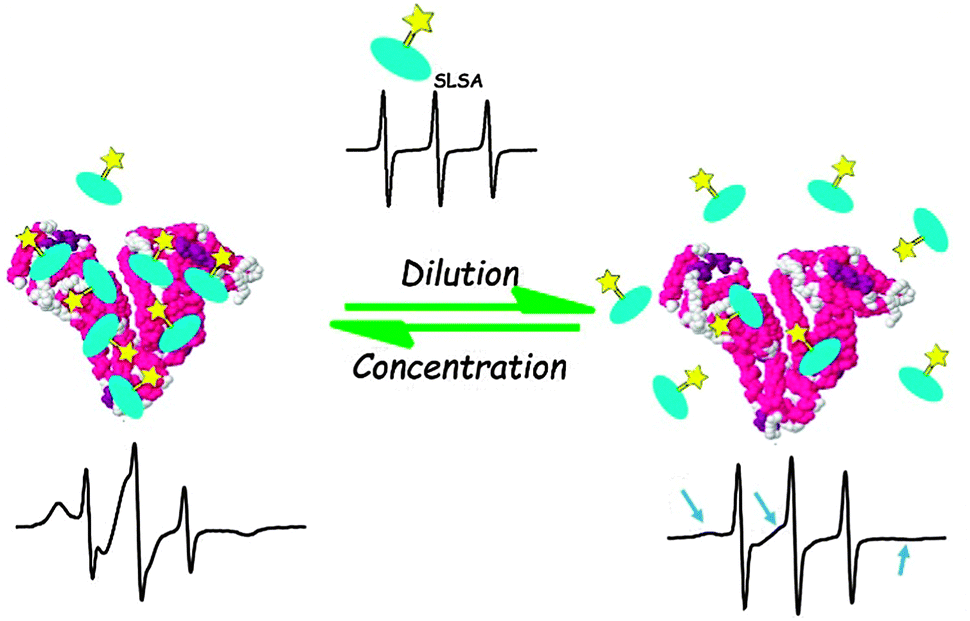 | ||
| Fig. 8 Sketch of SLSA binding to BSA at physiological concentrations (left) and at lower concentrations (<0.125 mM) (right) of BSA as derived from the cw EPR results. Spin labeled salicylic acids are shown with turquoise salicylic acid group and yellow Tempo group. The EPR spectrum of free SLSA is shown at top. | ||
SLSA release study from BSA
The SLSA release profile of BSA was also investigated at two temperatures 25 °C and 37 °C. 0.5 mM SLSA/BSA (1:1) solution placed in a D-tube dialyzer was measured by EPR spectroscopy as a function of time (Fig. 9). At time zero the fraction of bound SLSA is about 0.8. Therefore, two types of SLSA (bound and unbound) are present in the dialyzer tube. During the dialysis, EPR line intensities coming from both bound and unbound SLSA decrease together with time (Fig. 9A). This suggests that the amount of SLSA diffused through the membrane consists of the SLSA released from BSA and a portion of the unbound SLSA.
 | ||
| Fig. 9 (A) Cw EPR spectra of 0.5 mM SLSA/BSA (1:1) solution in a dialyzer tube as a function of time. (B) Normalized EPR spectra of (A) to the intensity of the high field line of the unbound SLSA. (C) The bound SLSA release profile of BSA with time at 25 °C and 37 °C. | ||
Expectedly, the relative diffusion rate of SLSA increases with temperature. At 37 °C an initial rapid release followed by a slower release was observed. Instead, a relatively steady release rate was observed at 25 °C. Nevertheless, the same amount of bound SLSA (92%) is released from BSA at the end of 28 hours both at 25 °C and 37 °C. In fact, while the total EPR intensity was gradually decreasing, the fraction of bound SLSA (0.8) did not change in the dialyzer tube (Fig. 9B).
We observe the bound and unbound SLSA signals together during the dialysis. This indicates the existence of an equilibrium between bound and unbound states.
| SLSA + BSA ⇔ SLSA–BSA complex |
Kinetics of the binding process
Drug affinity for protein binding sites can be judged by Ka (association constant), Kd (dissociation constant, 1/Ka) and ΔG (binding free energy). When the drug–albumin association constant is high, the drug releasing from albumin becomes very difficult. On the other hand, when the drug–albumin association constant is low, the drug loading becomes very weak and the drug is metabolized very quickly. Therefore, a critical range between strong and weak association is necessary for the drug transportation.The drug–albumin association constants at different concentrations of albumin were calculated using EPR results from Fig. 5B (Fig. S8 and Table S1, ESI†). The binding interaction between a SLSA and a BSA at equilibrium is described by the following:
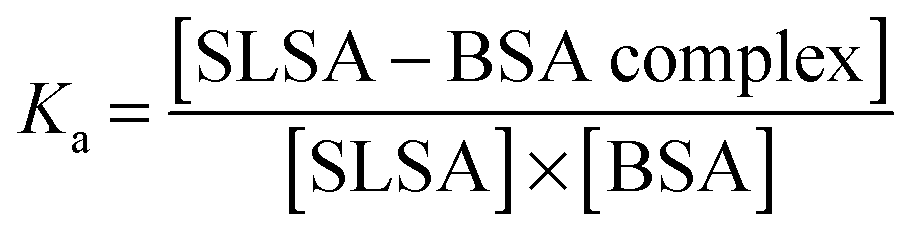 | (3) |
:1 ratio of initial [SLSA]:[BSA].
Drug immobilization on the protein is an entropically unfavorable process in an aqueous environment. Differences in the solvation free energies of drug and protein from the drug–protein complex provide the binding free energy of the drug with protein. However, the formation of energetically favorable weak bindings (electrostatic interactions, hydrophobic forces, van der Waals interactions, and hydrogen bonds) between the drug and protein compensates it and leads to increased Ka values. Binding free energy (ΔG) of SLSA with BSA includes noncovalent interactions, and so it can be related to the binding affinity by the following:
| ΔG = −RTlnKa | (4) |
:BSA, 1:1).
Fig. S8 (ESI†) shows that at physiological concentrations the Ka values are about 3.7–4.8 × 104 M−1. Below the physiological concentrations of albumin the Ka values decrease to about 2.0 × 104 M−1. Above the physiological concentrations of albumin, first the Ka values remain constant but at a high concentration for example 1.5 mM of BSA the Ka value reaches 9.0 × 104 M−1. These different Ka values show that at physiological concentrations of albumin, SLSA is loaded by albumin fairly but not so strongly which is crucial for the drug transportation.
Conclusions
Here, we report an application of cw EPR spectroscopy to investigate the drug binding to and release from serum albumin. Spin labeling of a diamagnetic drug allows monitoring of the drug directly. Therefore, we use Tempo based radicals to label the SA drug, and measure the SLSA in the presence of BSA. In the aqueous solution, the cw EPR spectroscopy clearly distinguishes whether SLSA is bound to BSA or not due to its sensitivity to the rotational dynamics of the SLSA.In summary, increasing the albumin concentration, increases the bound fraction of SLSA. Interestingly, the drug affinity of albumin reaches the saturation point just above the physiological concentration range (0.5–0.7 mM) of albumin. Moreover, albumin loses its affinity for the SLSA rapidly just below the physiological concentration range. Therefore, even a small decrease in albumin concentration under pathological conditions may produce a large increment in the amount of unbound drug which may lead to toxicity.
The binding stoichiometry of SLSA to BSA was found to be different depending on the concentration of BSA. The loading capacity of BSA increases with increasing concentration of BSA. At physiological concentration of BSA (0.5 mM), the maximum loading capacity of one BSA molecule is about 7 SLSA molecules. Whereas, for the diluted BSA concentrations (<0.125 mM) maximum ∼2 SLSA can bind to one BSA molecule.
Finally, EPR data show the reversible binding of SLSA to BSA. In the dialysis experiment, the EPR signal intensities of both bound and unbound SLSA decrease continuously while the ratio of bound to unbound SLSA concentrations remains constant. This suggests that the bound fraction of SLSA remains constant as long as the concentration of BSA does not change.
Acknowledgements
We thank Dr Gülçin Çakan-Akdoğan and Dr Mustafa Emrullahoğlu for helpful discussions. This work was financially supported by Turkish Scientific and Technological Research Council (Tubitak) via 2232 Program under grant 114C082.References
- J. Shen, S. Wanwimolruk, R. D. Purves, E. G. McQueen and M. S. Roberts, J. Pharmacokinet. Biopharm., 1991, 19, 575 CrossRef CAS PubMed
.
-
K. D. Rainsford, Aspirin and Related Drugs, Taylor & Francis, London, 2004 Search PubMed
- M. A. Zeitlinger, H. Derendorf, J. W. Mouton, O. Cars, W. A. Craig, D. Andes and U. Theuretzbacher, Antimicrob. Agents Chemother., 2011, 55, 3067 CrossRef CAS PubMed
- W. R. Ravis, D. L. Parsons and S. J. Wang, J. Pharm. Pharmacol., 1988, 40, 459 CrossRef CAS PubMed
- Y. Ni, R. Zhu and S. Kokot, Analyst, 2011, 136, 4794 RSC
- S. Nafisi, G. B. Sadeghi and A. Panahyab, J. Photochem. Photobiol., B, 2011, 105, 198 CrossRef CAS PubMed
- M. Maciazek-Jurczyk, A. Sulkowska, B. Bojka, J. Rownicka-Zubik and W. W. Sulkowski, Spectrochim. Acta, Part A, 2011, 82, 181 CrossRef CAS PubMed
- K. A. Majorek, P. J. Porebski, A. Dayal, M. D. Zimmerman, K. Jablonska, A. J. Stewart, M. Chruszcz and W. Minor, Mol. Immunol., 2012, 52, 174 CrossRef CAS PubMed
- G. Martini and L. Ciani, Phys. Chem. Chem. Phys., 2009, 11, 211 RSC
- S. Kempe, H. Metz and K. Mader, Eur. J. Pharm. Biopharm., 2010, 74, 55 CrossRef CAS PubMed
- N. Rapoport and L. Pitina, J. Pharm. Sci., 1998, 87, 321 CrossRef CAS PubMed
- H. Omote and M. K. Al-Shawi, J. Biol. Chem., 2002, 277, 45688 CrossRef CAS PubMed
- A. Besheer, K. M. Wood, N. A. Peppas and K. Mader, J. Controlled Release, 2006, 111, 73 CrossRef CAS PubMed
- W. Flores-Santana, T. Moody, W. Chen, M. J. Gorczynski, M. E. Shoman, C. Velazquez, A. Thetford, J. B. Mitchell, M. K. Cherukuri, S. B. King and D. A. Wink, Br. J. Pharmacol., 2012, 165, 1058 CrossRef PubMed
- Y. Nagasaki, Ther. Delivery, 2012, 3, 1 CrossRef
- W. L. Hubbell, D. S. Cafiso and C. Altenbach, Nat. Struct. Biol., 2000, 7, 735 CrossRef CAS PubMed
- Y. Akdogan and D. Hinderberger, J. Phys. Chem. B, 2011, 115, 15422 CrossRef CAS PubMed
- Y. Akdogan, M. J. N. Junk and D. Hinderberger, Biomacromolecules, 2011, 12, 1072 CrossRef CAS PubMed
- D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 153 CrossRef CAS PubMed
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1975, 11, 824 CAS
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38 CrossRef CAS PubMed
- A. A. Bhattacharya, T. Grüne and S. J. Curry, Mol. Biol., 2000, 303, 721 CrossRef CAS PubMed
- S. Park, B. L. Hayes, F. Marankan, D. C. Mulhearn, L. Wanna, A. D. Mesecar, B. D. Santarsiero, M. E. Johnson and D. L. Venton, J. Med. Chem., 2003, 46, 936 CrossRef CAS PubMed
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed
- F. Yang, C. Bian, L. Zhu, G. Zhao, Z. Huang and M. Huang, J. Struct. Biol., 2007, 157, 348 CrossRef CAS PubMed
- P. Bourassa, C. D. Kanakis, P. Tarantilis, M. G. Pollissiou and H. A. Tajmir-Riahi, J. Phys. Chem. B, 2010, 114, 3348 CrossRef CAS PubMed
- Y. J. Hu, Y. Ou-Yang, C. M. Dai, Y. Liu and X. H. Xiao, Biomacromolecules, 2010, 11, 106 CrossRef CAS PubMed
- D. Hinderberger, Top. Curr. Chem., 2012, 321, 67 CrossRef CAS PubMed
- J. A. Roberts, F. Pea and J. Lipman, Clin. Pharmacokinet., 2013, 52, 1 CrossRef CAS PubMed
- C. Bertucci and E. Domenici, Curr. Med. Chem., 2002, 9, 1463 CrossRef CAS PubMed
- Y. Ni, S. Su and S. Kokot, Anal. Chim. Acta, 2006, 580, 206 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Details of any supplementary information available should be included here. See DOI: 10.1039/c5cp03583j |
| This journal is © the Owner Societies 2015 |