
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lipopeptides: from self-assembly to bioactivity
Ian W.
Hamley
*
Dept of Chemistry, University of Reading, Whiteknights, Reading RG6 6AD, UK. E-mail: I.W.Hamley@reading.ac.uk
First published on 18th March 2015
Abstract
This Feature Article discusses several classes of lipopeptide with important biomedical applications as antimicrobial and antifungal agents, in immune therapies and in personal care applications among others. Two main classes of lipopeptide are considered: (i) bacterially-expressed lipopeptides with a cyclic peptide headgroup and (ii) linear lipopeptides (with one or more lipid chains) based on bio-derived and bio-inspired amino acid sequences with current clinical applications. The applications are briefly summarized, and the biophysical characterization of the molecules is reviewed, with a particular focus on self-assembly. For several of these types of biomolecule, the formation of micelles above a critical micelle concentration has been observed while others form bilayer structures, depending on conditions of pH and temperature. As yet, there are few studies on the possible relationship between self-assembly into structures such as micelles and bioactivity of this class of molecule although this is likely to attract further attention.
Introduction
Lipopeptides are a remarkable class of self-assembling molecule that are able to form peptide-functionalized supramolecular nanostructures. Lipopeptides are amphiphilic molecules which incorporate one or more lipid chains attached to a peptide headgroup. Self-assembly is observed depending on the hydrophile/lipophile balance of the molecules as well as interactions between the peptide units. The self-assembly of synthetic bio-mimetic and bio-inspired lipopeptides has been the subject of previous reviews and this is not covered again here.1 We present an up-to-date overview on the self-assembly of bio-derived and bio-active lipopeptides, with proven activity in on-the-market applications in medicine and other fields such as crop protection.Compared to the self-assembly of lipids, that of lipopeptides is relatively unexplored. This is remarkable, given their relevance to important applications in biomedicine, including use as last resort antimicrobial agents to treat for example MRSA (methicillin-resistant S. aureus) infections (e.g. daptomycin, Fig. 1),2 use in immune disease therapies (e.g. Pam3CSK4, Fig. 13)3 and in cosmeceuticals (e.g. Matrixyl™, C16-KTTKS).4 Other applications of lipopeptides extend beyond human medicine, including use as fungicides for crop treatment (e.g. surfactin, Fig. 7b),5 avoiding adverse environmental effects associated with a conventional synthetic pesticides.5b,6 For potential biomedical applications, lipopeptides offer advantages compared to peptides of improved amphiphilicity and compatibility with the lipid wall of cell membranes, enabling for example the delivery of actives into cells via endocytosis (vide infra). In addition, the self-assembly of lipopeptides facilitates the presentation of peptide functionalities at high density on the surface of nanostructures such as fibrils, micelles and vesicles.
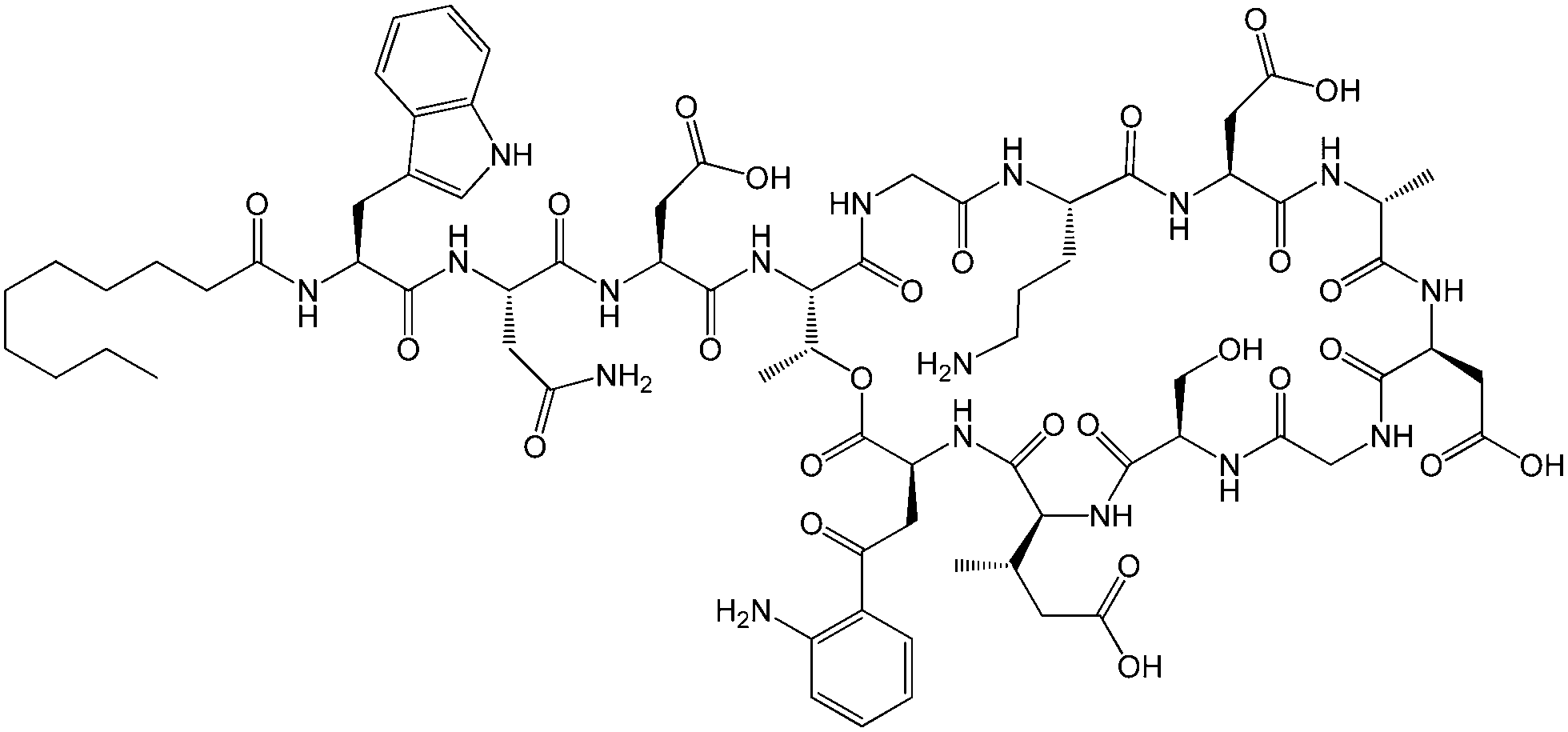 | ||
| Fig. 1 Molecular structure of daptomycin.11 | ||
In the main, naturally expressed bioactive lipopeptides contain a cyclic peptide headgroup attached to a single lipid chain. Cyclisation of the peptide unit as in many bacterially expressed lipopeptides enhances the in vivo stability of the motif compared to the linear counterpart (and thus can be expected to have evolved to be expressed in the cyclic form). This is due to reduced proteolysis resulting from the absence of free C- and N-termini.7 Of course, cyclisation also restricts the peptide conformation which may also be relevant to bioactivity. Expressed lipopeptides usually comprise molecules with a range of lipid chain lengths (typically in the range C14–C18) and conformations. Another class of bioactive lipopeptides is usually produced synthetically based on bio-derived sequences. These lipopeptides often contain a linear peptide headgroup attached to one, two or three hexadecyl (palmitoyl) lipid chains. In the following we use the notation C16 or Pam for a hexadecyl lipid chain, depending on conventional usage for the relevant lipopeptide.
This review is focussed on lipopeptides, and does not cover the fascinating field of lipoproteins which also have a variety of in vivo biological roles, for example apolipoproteins are the major component of high density lipoproteins which are a risk factor associated with cardiovascular disease. The structure of apolipoproteins has been reviewed elsewhere.8 This is a mini-review aiming to introduce the main classes of bioactive lipopeptides investigated to date, relating their structure to activity and self-assembly properties where these have been investigated (which is only the case for a few materials). It is not our purpose to exhaustively review all studies on the biological activity of lipopeptides, of which there are many hundreds published. We apologise to the authors of papers whose work has not been included.
Previous reviews have been published covering lipopeptides as a class of biosurfactant.9 In addition, reviews focussed specifically on the natural functions of lipopeptides from Bacillus and Pseudomonas species are available.6b,10
As discussed in the following, lipopeptides may self-assemble into spherical micelles above a critical micelle concentration (cmc) or into other structures (especially nanofibrils and nanotapes), in which case we use the notation critical aggregation concentration (cac) to denote the concentration above which self-assembly is observed.
Bacterially expressed cyclic lipopeptides
Lipopeptides are expressed by many bacteria, having several roles including activity against other microorganisms including bacteria, fungi and viruses, involvement in bacterial motility and swarming behaviour and in attachment to surfaces.6b,10e The host defense mechanism produces lipopeptides in response to other microorganisms including fungi, viruses, mycoplasma and bacteria. Two genera of bacteria, Bacillus and Pseudomonas have produced lipopeptides that have been particularly extensively studied. They have important applications due to antibacterial, antifungal and antiviral activity as discussed in the following.Daptomycin (Fig. 1) is a lipopeptide comprising a decanoyl lipid chain attached to a partly cyclised 13-amino acid peptide. Daptomycin is produced by the Gram-positive bacterium Streptomyces roseoporous.12 It has potent antimicrobial properties and is clinically approved for use as an antibiotic for serious infections caused by Gram-positive bacteria such as MRSA (methicillin-resistant Staphylococcus aureus) and vancomycin-resistant enterococci.12a It is marketed under the tradename Cubicin by Cubist Pharmaceuticals2c,13 (recently acquired by Merck). It incorporates two non-natural amino acids: L-kynurenine, which is unique to daptomycin, and L-3-methylglutamic acid. The former residue, containing an aniline unit, gives rise to interesting fluorescence properties, in particular an emission peak at 460 nm.14 The mode of activity of daptomycin is still unclear.12a,15 One hypothesis involves the inhibition of lipoteichoic acid synthesis (lipoteichoic acid is a proteoglycan which is a major component of the cell wall of Gram-positive bacteria).15 Another proposed mechanism involves disruption of bacterial membrane potential, for example via pore formation.15 The latter mechanism seems favoured based on recent work examining ion-dependent pore formation in model membranes,16 along with the fact that its activity is calcium ion dependent.17
The existence of a critical aggregation concentration (cac) has been determined for daptomycin by making use of the afore-mentioned fluorescence properties of the lipopeptide, as well as NMR and light scattering techniques.14b The cac has been measured as a function of pH, temperature and calcium ion concentration.14b The aggregation number in aqueous solution at pH 4–6.5 is reported as (18 ± 2). Fig. 2 shows a cryo-TEM recently obtained by our group showing spherical micelles formed by daptomycin.
 | ||
| Fig. 2 Cryo-TEM image of micelles formed by daptomycin (2.5 mM in 0.625 mM CaCl2).18 | ||
The echinocandins are a class of cyclic lipopeptide compounds with antifungal activity. Examples of this class of molecule are shown in Fig. 3. The most widely known is caspofungin, marketed as a drug by Merck and Co. The antifungal activity of this class of compound is due to the interaction of lipopeptide molecules with the insoluble polysaccharide component of the cell wall of fungal cells, specifically due to the inhibition of the synthesis of glucans.19 The interactions of echinocandins with Candida fungi such as C. albicans has been particularly well studied.19,20 The self-assembly properties of these compounds seem to be largely unexamined although Eli Lilly have patented micellar formulations of echinocandins with conventional surfactants.21
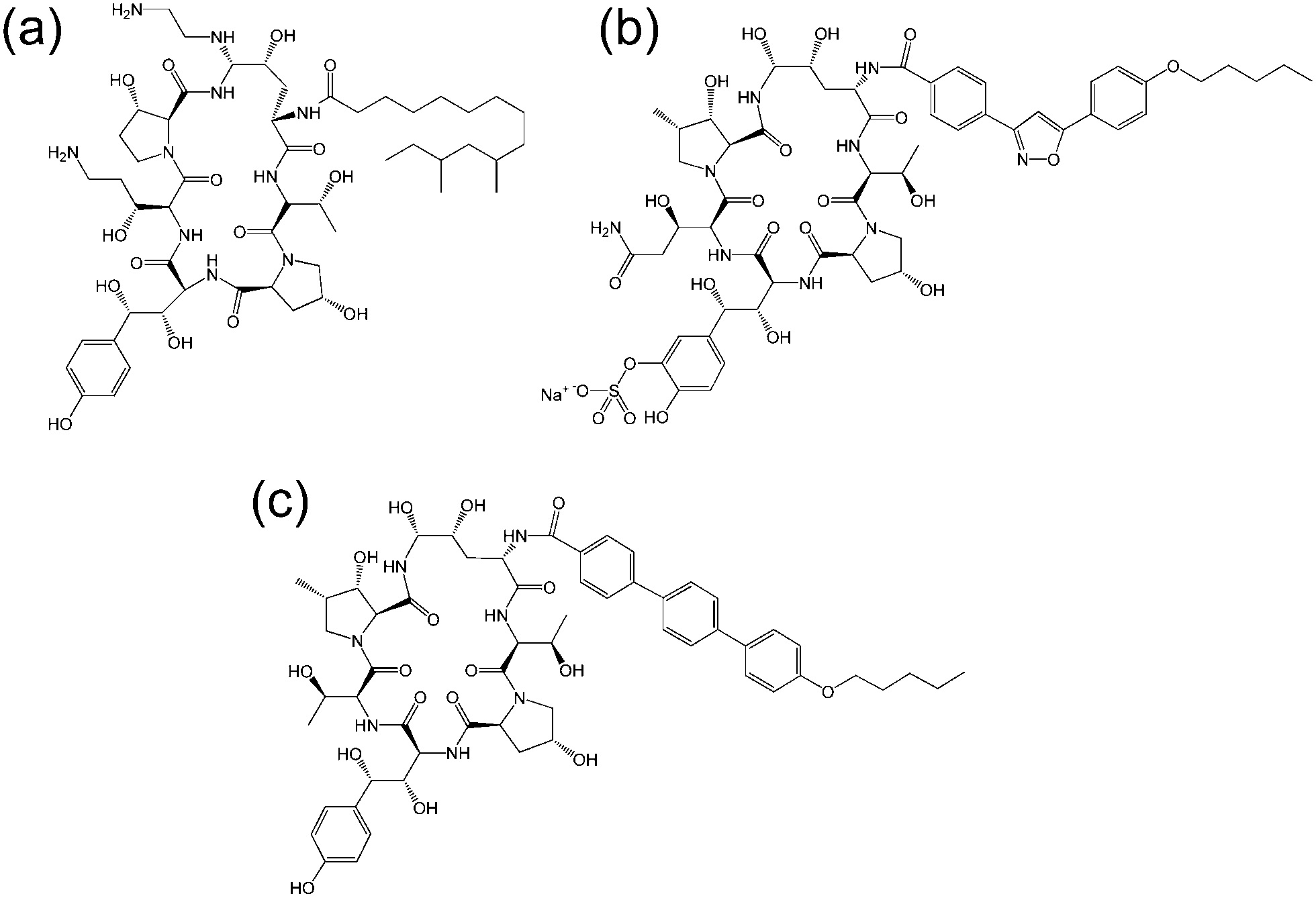 | ||
| Fig. 3 Molecular structure of several echinocandins. Adapted from ref. 19. (a) Caspofungin, (b) micafungin, (c) anidulafungin. | ||
Viscosin (Fig. 4) is a cyclic lipopeptide obtained from Pseudomonas libanensis23 or Pseudomonas fluorescens.22 It has antibiotic activity as shown against the tubercle bacillus.24 Viscosin is highly surface active23a,25 and is able to inhibit the migration of cancer cells.23bFig. 5 shows surface tension data used to determined a cmc = 0.15 mg ml−1 for viscosin in aqueous solution. Viscosin and the related cyclic lipopeptide massetolide (Fig. 4) also produced by P. fluorescens are able to protect it from protozoan predation.22
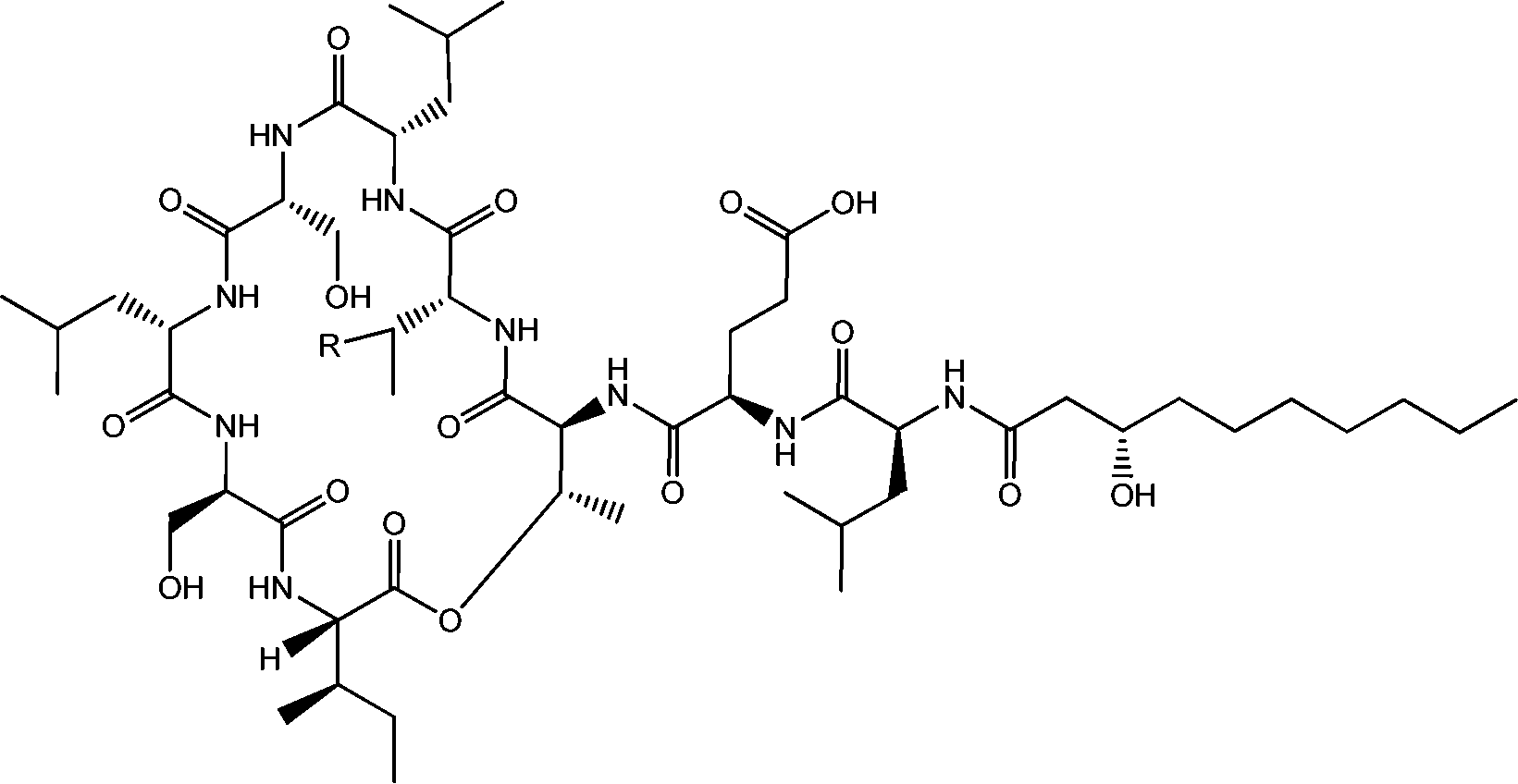 | ||
| Fig. 4 Viscosin (R = H) and massetolide (R = CH3).22 | ||
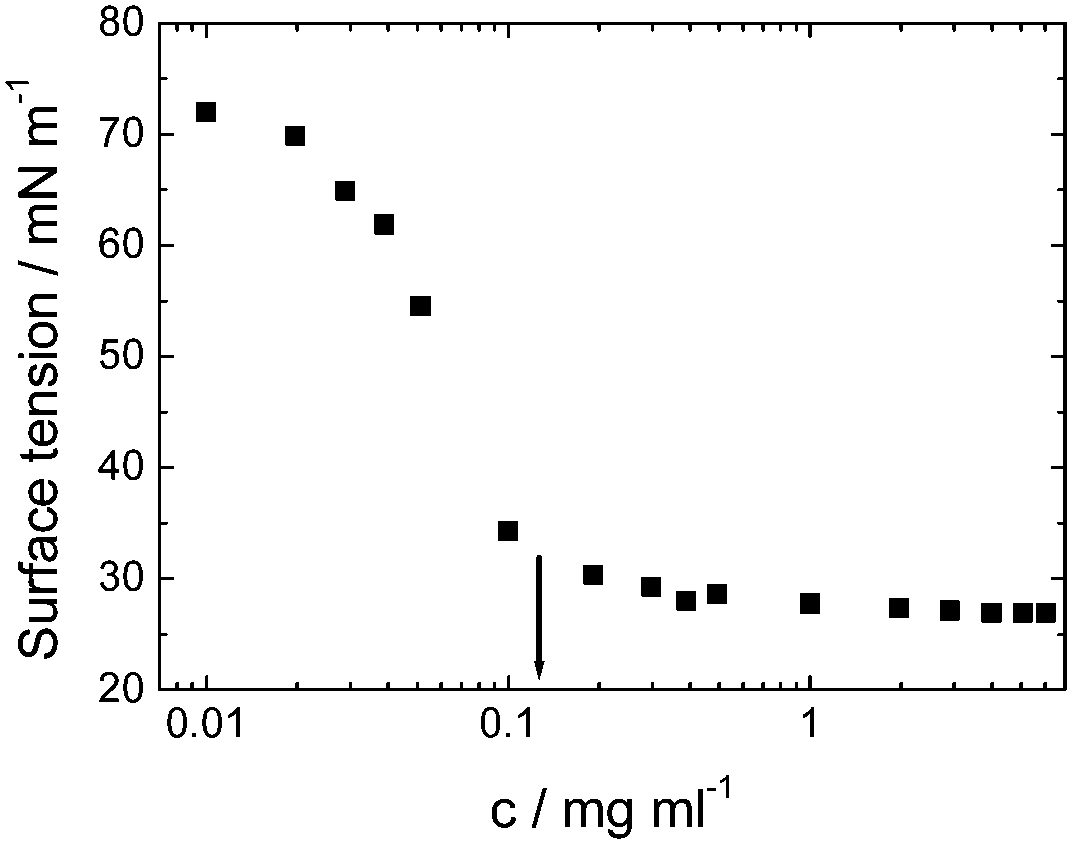 | ||
| Fig. 5 Surface tension versus concentration used to determine the cmc for viscosin (shown by arrow). Redrawn from ref. 23a. | ||
Lichenysins,27 pumilacidins28 and polymyxin B (Fig. 6)29 are classes of antimicrobial lipopeptides produced by Bacillus licheniformis, Bacillus pumilus and Bacillus polymyxa respectively. Lichenysin A (Fig. 7a) is a powerful surfactant, and antimicrobial activity against several Gram positive and Gram negative bacteria, although not as great as that observed for surfactin.27a Several pumilacidins show antiviral activity against herpes simplex virus as well as anti-ulcer activity.28 Polymyxin B (Fig. 6) and polymyxin E (colistin) show antibiotic activity against a range of Gram-negative bacteria.29 The two molecules are distinguished by the substitution of a D-leucine in colistin with a D-phenylalanine in polymyxin B.26b The mode of action of these lipopeptides has been proposed to be membrane disruption due to interaction between the cationic polymyxin and the anionic bacterial outer membrane leading to a detergent-like activity.29
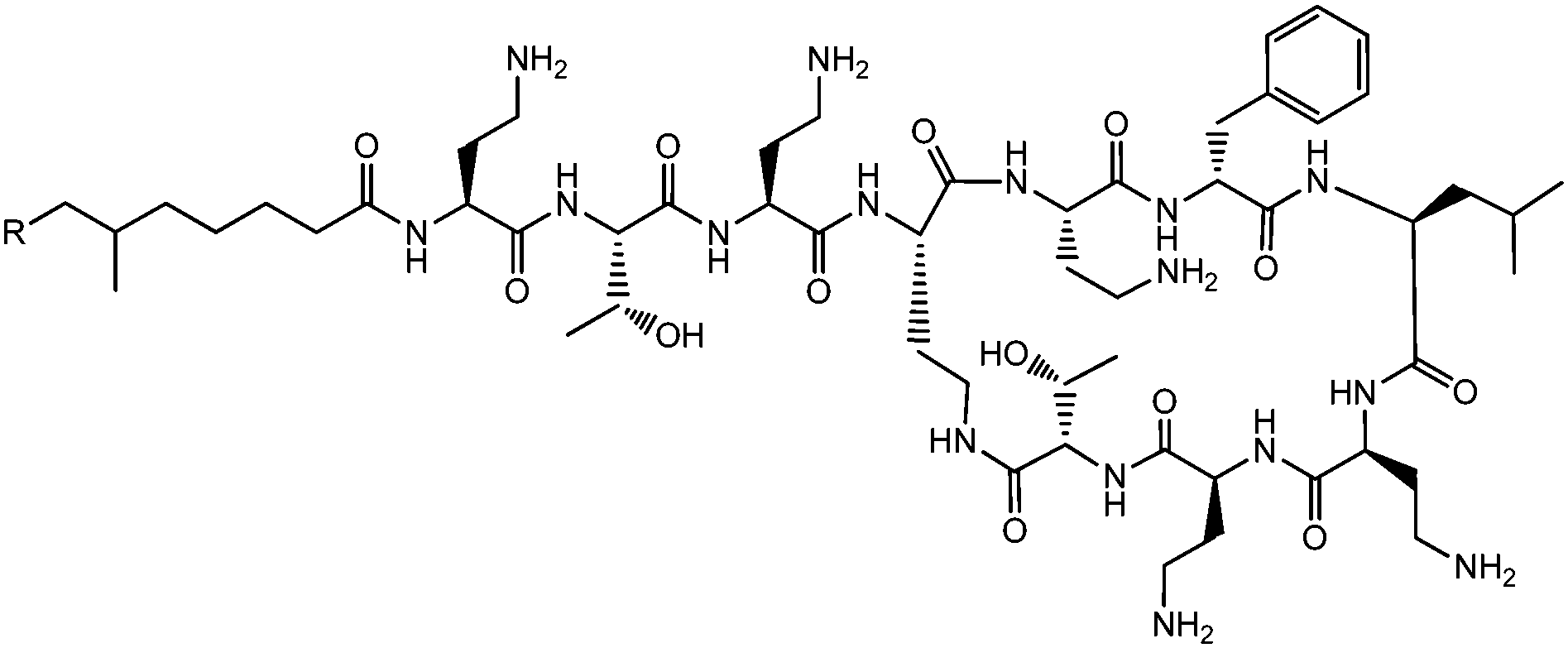 | ||
| Fig. 6 Structure of polymyxin B.26 Polymyxin B is a mixture of lipopeptides with different lipid chain lengths (here indicated by R). | ||
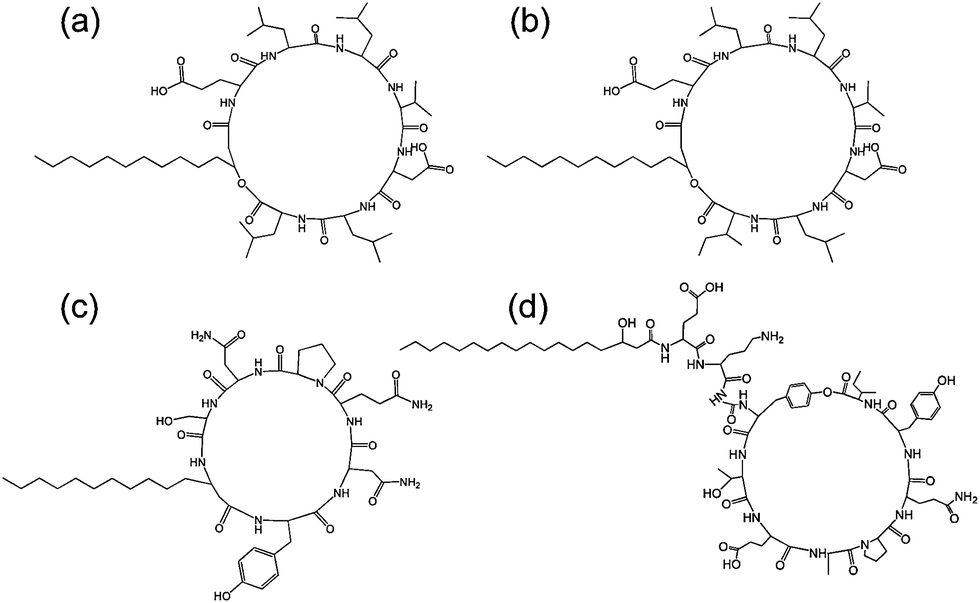 | ||
| Fig. 7 Structure of some lipopeptides produced by B. subtilis. (a) Surfactin (b) lichenysin A, (c) iturin A, (d) fengycin/plipastatin A. Redrawn from ref. 5b. As detailed in ref. 5b, the expressed lipopeptides actually comprise a mixture of lipid chain lengths. | ||
Fig. 7 shows the structure of several lipopeptides produced by B. subtilis.30 Variants of surfactins, iturins and lichenysins can be genetically engineered to incorporate different amino acids (via for example, incorporation in the bacterial culture medium). Synthetic variants have also been examined. The engineered/synthetic Bacillus lipopeptides have distinct structure–activity relationships as reviewed elsewhere.5a,b,10b All of these classes of lipopeptide are potent antifungal agents with applications in crop protection.6a,31 The effect of surfactin on the swarming behaviour of B. subtilis has been examined, its absence reducing swarming activity.32
Cyclic lipopeptides produced by Pseudomonas species include the viscosin group (as discussed above), the amphisin group, the syringomycin group and the tolaasin group (Fig. 8).10e,33 The biosynthesis of these lipopeptides is discussed elsewhere.10e There are few studies on the aggregation behaviour of these lipopeptides although a report on the biosurfactant and ion-channel forming properties of two lipopeptides from the syringomycin group is available.34 This includes a cmc measurement of 0.8 mg ml−1 for both syringomycin and syringopeptin from a Pseudomonas syringae strain. Calcium ion channel formation was observed at nanomolar concentrations showing the cytotoxicity of these lipopeptides to plant cells.34
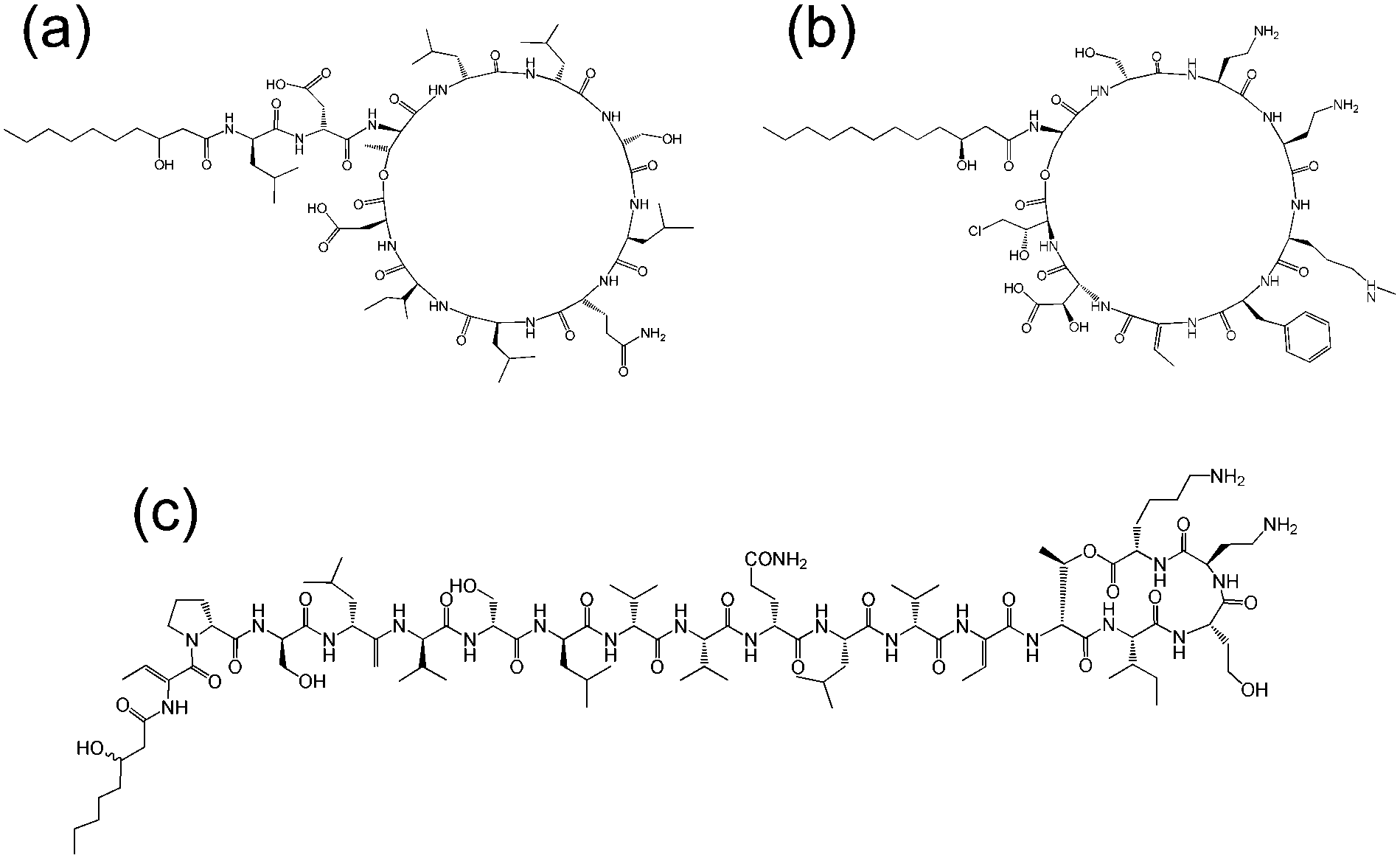 | ||
| Fig. 8 Molecular structures of lipopeptides produced by Pseudomonas species. (a) Amphisin35 (b) syringomycin,36 (c) tolaasin I.37 | ||
Surfactin is a highly surface-active biosurfactant reducing the surface tension of water to 27 mN m−1 at a concentration as low as 20 μM.10b The cmc at room temperature is 7.5 μM. The temperature dependence of the cmc has been examined, along with the thermodynamic parameters of micellization, via isothermal titration calorimetry.38 The cmc of surfactin is very low compared to that of many surfactants such as non-ionic surfactants of the CnEOm class where EO denotes ethylene oxide and the subscripts are the number of repeats. This is shown in Fig. 9 which plots the water/phospholipid bilayer partition coefficient against cmc.38 The partition coefficient, K, was analysed as part of a study on membrane permeabilization. There is also a relationship between the surfactant-to-lipid mole ratio at the onset of solubilisation and K × cmc.38
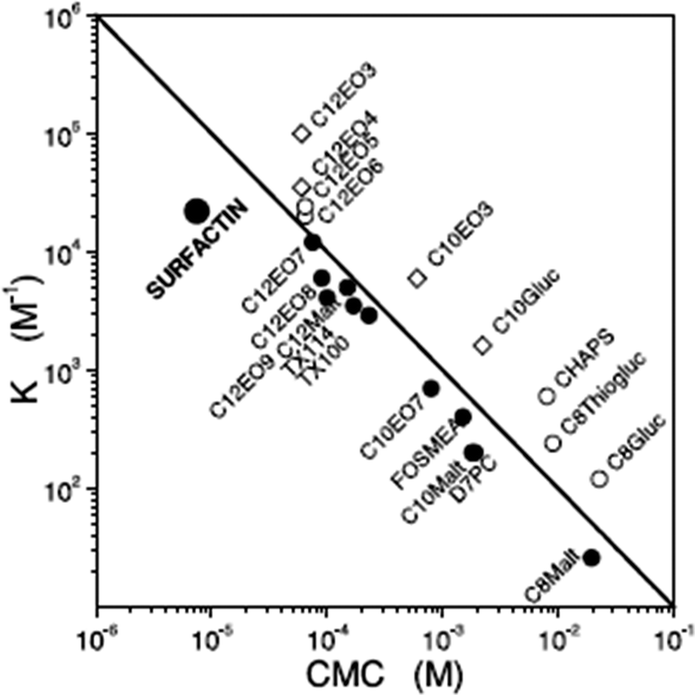 | ||
| Fig. 9 Relationship between the water/phospholipid partition coefficient K and the cmc for surfactin compared to several types of non-ionic and ionic surfactant. CnEOm are alkyl ethyoxylate non-ionic surfactants, CnMalt, CnGluc and CnThiogluc are alkyl maltosides, glucosides or thioglucosides, TX denotes triton ethylene oxide-based non-ionic surfactants, CHAPS is a steroid, D7PC is diheptanoylphosphatidylcholine, and FOSMEA is dodecyl phospho-n-methylethanolamine.38 The line corresponds to K × cmc = 1. An offset from the diagonal to lower K values indicates strong membrane destabilization by the respective compound. | ||
The aggregation of surfactin (Fig. 7a) in bulk solution and at air/water and hydrophobic solid/water interfaces was investigated using neutron scattering techniques.39 Small-angle neutron scattering indicated the presence of small (association number p = 20) spherical micelles in bulk aqueous solution whilst at the interface (either air/water or silane/water) surfactin was found to adopt a globular conformation.39 Neutron reflectometry was also used to probe the interaction between surfactin and solid-supported phospholipid bilayers – below the cmc of surfactin a mixed bilayer was reported.40 Later, as part of a study also including other Bacillus subtilis lipopeptides, the self-assembly of surfactin in aqueous solution was investigated via cryogenic transmission electron microscopy (cryo-TEM) and small-angle X-ray scattering (SAXS), and the secondary structure was probed using circular dichroism (CD) and FTIR spectroscopic methods.30 Cryo-TEM and SAXS revealed spherical micelles with a radius of 2.0 nm, consistent with the findings of Shen et al.39 but contradictory to the report of Ishigami et al.41 who reported large rod-shaped micelles, albeit in a different buffer (NaHCO3). CD and FTIR were consistent with random coil and turn structures for surfactin (again contrary to Ishigami et al. who reported β-sheet secondary structure41). Similar findings in regard to self-assembled nanostructure and secondary structure were noted for plipastatin (Fig. 7d). Unexpectedly, and in complete constrast to surfactin and plipastatin, mycosubtilin shows a distinct self-assembly motif – extended nanotapes based on bilayers of lipopeptide molecules.30 This difference may be due to branching in the lipid chains of mycosubtilin and/or due to conformational differences in the cyclic peptide headgroup, although plipastatin also includes backbone and side-chain aromatic units.
The critical micelle concentration of iturin A (Fig. 7c) was determined by fluorescence assays using the intrinsic fluorescence of the D-tyrosyl residue (which is common to all iturins10a) leading to a value of 25–40 μM in water.10a,42 The distribution of conformers was found to be influenced by the cmc. The interaction of the lipopeptide with model (egg phosphatidylcholine) vesicles was investigated: above the cmc, fluorescence revealed self-association of iturin A inside the lipid membranes, an effect not observed below the cmc.42 The micelle size and aggregation number have been estimated.25 Far above the cmc, TEM images suggest the presence of vesicles and an interdigitated bilayer structure was proposed for the vesicle walls.43 Iturin A has been shown to form potassium ion-conducting channels in lipid bilayers.10a,44
Surfactin is able to permeabilize lipid membranes,38,45 and it forms ion channels in planar lipid bilayer membranes.46 The interaction of surfactin with POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) vesicles has been investigated via isothermal titration calorimetry and this has been correlated to vesicle lysis probed by calcein fluorescence.47 Detergent-like permeabilization effects were observed on increasing concentration even below the cmc (in the range 7.5 μM to 10 μM depending on buffer38,45) although complete solubilisation and the formation of mixed micelles occurs at the cmc.45,47 The study by Carillo et al. suggests that membranes are permeabilized by dimers of surfactin.45
Surfactin has a wide range of demonstrated activities, including antifungal and antibiotic properties.9b,10b,c,d For the latter, a pore-formation mechanism was proposed based on permeabilization studies (using carboxyfluorescein as a fluorescent probe) using model lipid membranes.45 A range of other very useful activities have been reported9b,10c,d,48 including antiviral and antitumor properties and effectiveness as an anti-mycoplasma treatment. Surfactin has been shown to suppress inflammatory responses through inhibition of phospholipase A2.49 It is able to suppress biofilm formation by bacteria such as Salmonella enterica and this was examined in PVC plates as well as urethral catheters, pointing to relevance to applications in healthcare.50 Surfactin C can enhance plasminogen activation, and enhance fibrinolysis in vivo.51 This thrombolytic activity is relevant to conditions involving blood clotting including myocardial and pulmonary disorders. Linear analogues of surfactin have been shown to have reduced haemolytic activity compared to native cyclic variants. They were also observed to protect against lytic (Triton) surfactant-induced haemolysis.52 This was correlated to micellization, i.e. there is evidence for a relationship between self-assembly and bioactivity from this study. Two mechanisms for this were proposed – (i) the formation of mixed micelles of the lytic surfactant and the protective surfactin derivative, (ii) differences in the insertion of the two surfactants into the cell membrane.52
It has been reported that lichenysin (Fig. 7a) has a much lower cmc than surfactin (22 μM vs. 220 μM in 5 mM Tris buffer) and is also a better chelator of Mg2+ and Ca2+ ions.27b As noted above, the cmc of surfactin is very low in water and other buffers,38,45 and in fact the value 220 μM for surfactin seems too large compared to those reported previously (also in Tris buffer).
Immune response lipopeptides
A series of lipopeptides that act as Toll-like receptor (TLR) agonists have been developed, with important applications in the treatment of infectious diseases. TLRs are transmembrane proteins with a key role in the immune system and as such are important therapeutic targets to treat infections.53 A list of TLR targeting immuno-stimulatory agents being developed in animal models to treat various viral and bacterial infections is available.54 The toll-like receptor domains have been studied by X-ray crystallography and, upon binding to PamnCSK4 lipopeptides (vide infra) reveal remarkable horse-shoe structures resembling partial β-barrels (Fig. 10).55 The lipopeptide is bound to the outer surface of the complex.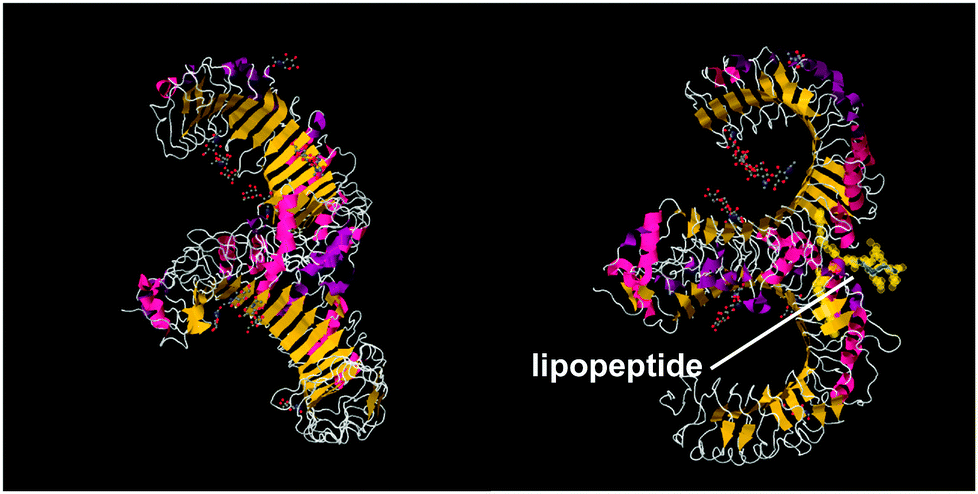 | ||
| Fig. 10 Two views of X-ray crystal structure for the TLR1/TLR2 complex bound by Pam3CSK4 (pdb file 2Z7X).56 The right-hand image shows the location of the bound lipopeptide molecule (represented as yellow spheres). | ||
Pam3Cys lipopeptides (Fig. 11) are synthetic versions of the N-terminal sequence of the principal lipoprotein of E. coli, also known as Braun's lipoprotein,57 that is abundant in the cell wall of Gram-negative bacteria.58 Pam3Cys-based lipopeptides are able to stimulate cytotoxic T lymphocyte (CTL) responses against influenza virus-infected cells (when linked to influenza nucleoprotein viral peptides).59 A related lipopeptide (Pam3CysSerSer linked to a foot-and-mouth disease virus protein sequence) is able to stimulate antibodies against foot-and-mouth disease.60 Pam3CysSerSer is able to mediate attachment to the cell membrane, internalization into the cytoplasm and to activate macrophages to secrete cytokines.59 As shown in Fig. 10, Pam3CSK4 binding leads to an m-shaped heterodimer of the TLR1 and TLR2 ectodomains, not observed for Pam2CSK4.56 A similar crystal structure is reported for Pam2CSK4 bound to the TLR2/TLR6 heterodimer.55
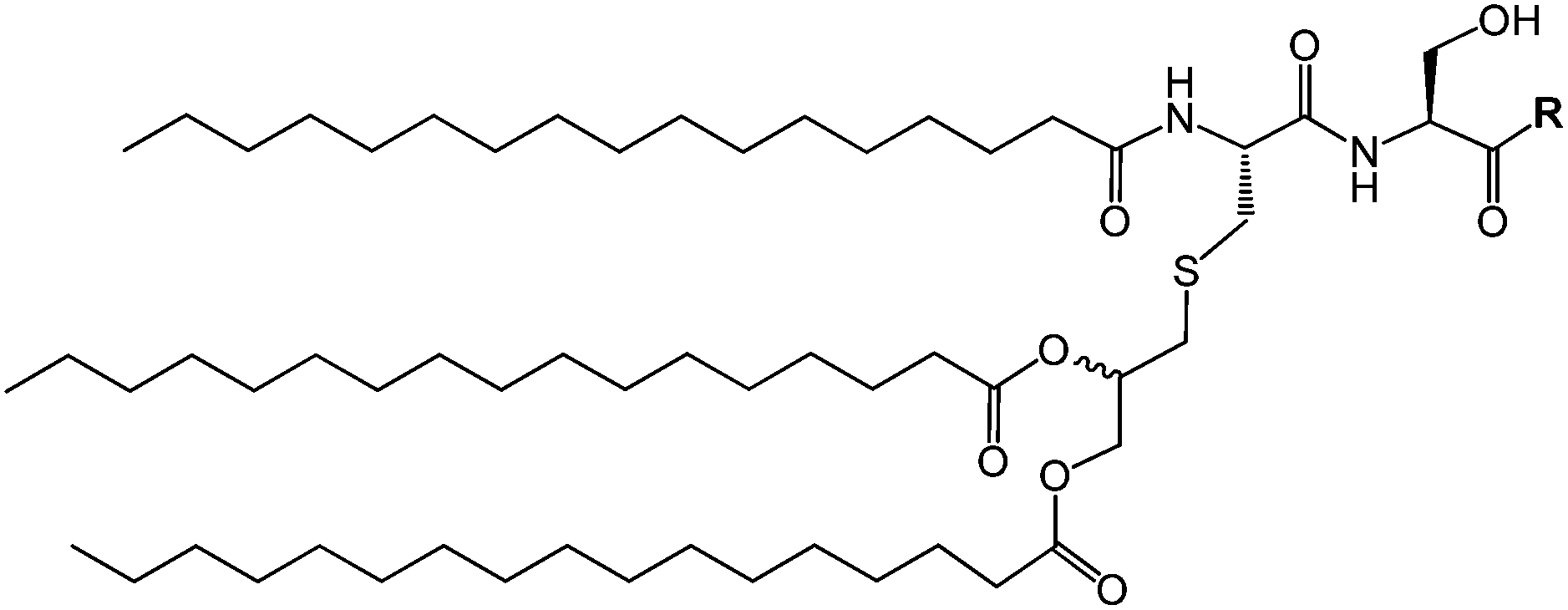 | ||
| Fig. 11 Scheme of generic Pam3Cys-based lipopeptide structure where R indicates a peptide sequence. | ||
The Pam2Cys homologue of Pam3Cys lipopeptides has been shown to correspond to the lipid moiety of macrophage-activating lipopeptide 2 from mycoplasma.61 The presence of only two lipid chains in Pam2Cys lipopeptides along with a free amino group leads to improved water solubility. MALP-2, where MALP stands for macrophage-activating lipopeptide, was developed based on this lipopeptide with an attached GNNDESNISFKEK sequence (Fig. 12).61a,62 This lipopeptide is potent in stimulating macrophages or monocytes at pM concentrations.62 MALP-2 activates epithelial cells through TLR6 and TLR2, but not TLR1 and TLR2.63 Indeed, TLR2 and TLR6 respond exclusively to lipopeptides possessing a diacylglycerol group.64 Pam3CSK4 is recognised by TLR1 and TLR6 and does not require TLR2.63,64b In addition, the R stereoisomer of MALP-2 and Pam2CSK4 and Pam3CSK4 are more active than the respective S stereoisomer. Together, these results indicates the important role of the chirality of the central carbon in the diacylglycerol group in TLR recognition.64b The role of specific functional groups at the C terminus in the TLR2 activity of Pam2Cys was examined which showed that a variant containing a 1,3-dihydroxypropyl moiety showed higher activity than the 1,2-dihydroxypropyl derivative.3b Pam2Cys has been incorporated in other compounds, for instance a PEGylated analogue was prepared to examine the effect on influenza immunity when administered without antigen.65 Attachment of synthetic branched peptides rich in terminal arginine or glutamic acid residues to Pam2Cys leads to enhanced protein immunogenicity due to binding of charged protein (BSA as model agonist) to the oppositely charged branched lipopeptide derivative.66 The immune response was studied using influenza infected mice.
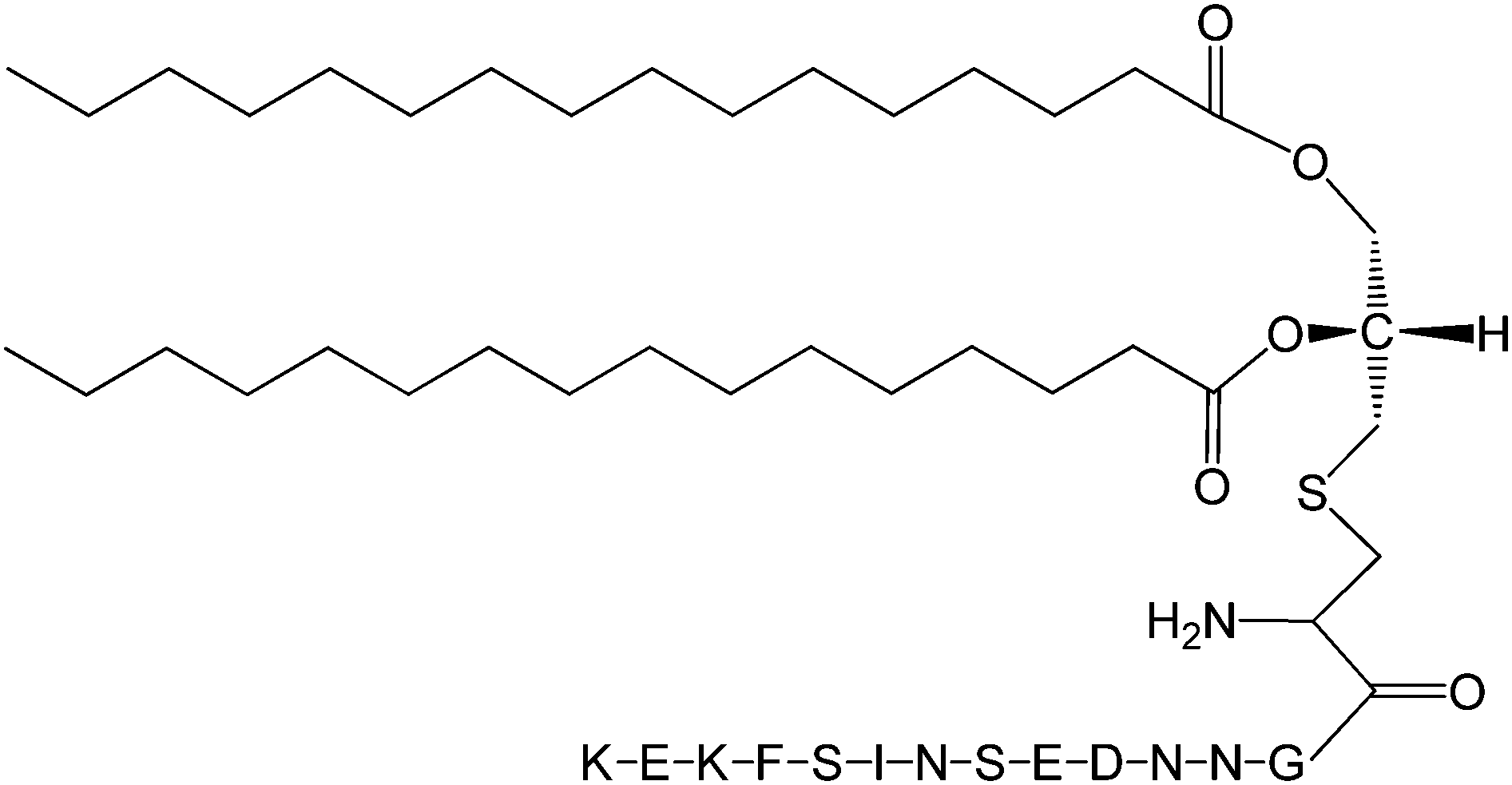 | ||
| Fig. 12 Molecular structure of MALP-2.61b | ||
Immunocontraceptive vaccines have been developed, incorporating a peptide sequence EHWSYGLRPG from luteinizing hormone-releasing hormone (LHRH).58 Lipopeptides incorporating LHRH are able to stimulate antibodies against this “self” hormone, leading to sterilization in a female mouse model. Lipopeptides were synthesized based around Pam3Cys and Pam2Cys templates incorporating the LHRH sequence which incorporates one or more epitopes for B cells (denoted [B]) as well as the CD4+ T cell epitope GALNNRFQIKGVELKS (denoted [T]) from the L chain of influenza virus hemagglutinin.58 Lipopeptides with different architectures were examined, branched lipopeptides with [T] and [B] attached peripherally, such as [T]-Lys-Pam2Cys-Ser-Ser-[B] induced a higher antibody titer than that observed for linear lipopeptides with [T]–[B] at the C terminus. In general, Pam2Cys-containing lipopeptides were stated to be better immunogens that Pam3Cys ones.58 It has recently been shown that monoacyl PamCys lipopeptides may be conveniently prepared via direct thiol–ene coupling, enabling to the preparation of self-adjuvating antigenic lipopeptides with TLR2 agonist activity.67 A truncated sequence from a cytomegalovirus (CMV) protein was used a model epitope as it is able to stimulate CD8+ cytotoxic T-cells.
Lipopeptide-based vaccines have been developed to treat several diseases including HIV,68 hepatitis B69 and HPV (human papilloma virus)70 infections (HPV is also implicated in several types of cancer including cervical cancer).
We recently investigated the self-assembly properties of three PamnCSK4 lipopeptides (n = 1–3) (Fig. 13).71 A remarkable dependence of morphology on the number of attached hexadecyl lipid chains was found, with spherical micelle structures for mono- and di-lipidated structures, but flexible wormlike micelles for the homologue containing three lipid chains. Images from cryogenic transmission electron microscopy (cryo-TEM) are shown in Fig. 14. The self-assembled structure was also confirmed by in situ small-angle X-ray scattering (SAXS) which enables the dimensions of the nanostructures to be determined. To investigate the thermal stability of the secondary structures, and the possible role of lipid chain melting, we performed temperature-dependent CD measurements. Neither PamCSK4 nor Pam2CSK4 show any significant change in the disordered conformation in the temperature range 20–60 °C which covers palmitoyl lipid chain melting temperatures, i.e. the lipid chain melting transition previously observed for palmitoyl lipopeptides.72 In contrast, Pam3CSK4 exhibits a discontinuity in spectra between 30 °C and 40 °C, from a β-sheet conformation at lower temperature to disordered at high temperature. This transition is partially reversible. We associate this transition with the lipid chain melting transition within the tri-palmitoyl chain lipopeptide. The transition from the lipid gel to sol state (at around 40 °C) has previously been shown to change the nanostructure of self-assemblies (twisted ribbons or tapes) of gemini C16 surfactants.73 We suggested that the distinct modes of assembly may have an important influence on the bioactivity of this class of lipopeptide. Interestingly, SAXS on more concentrated samples (5 wt% lipopeptide) indicates that Pam2CSK4 and Pam3CSK4 form complex cubic structures.74 In the same study, a lipolanthionine peptide (2R,6R)-Pam3LanHdaSer(Lys)4-NH2 [Lan: lanthionine, Hda: hexadecanoic acid] was found to form a lamellar phase (this lipopeptide in contrast to the PamnCSK4 compounds exhibits an inhibitory effect on activation of human macrophages).74 We are currently investigating the lyotropic polymorphism of PamnCSK4 compounds in more detail.
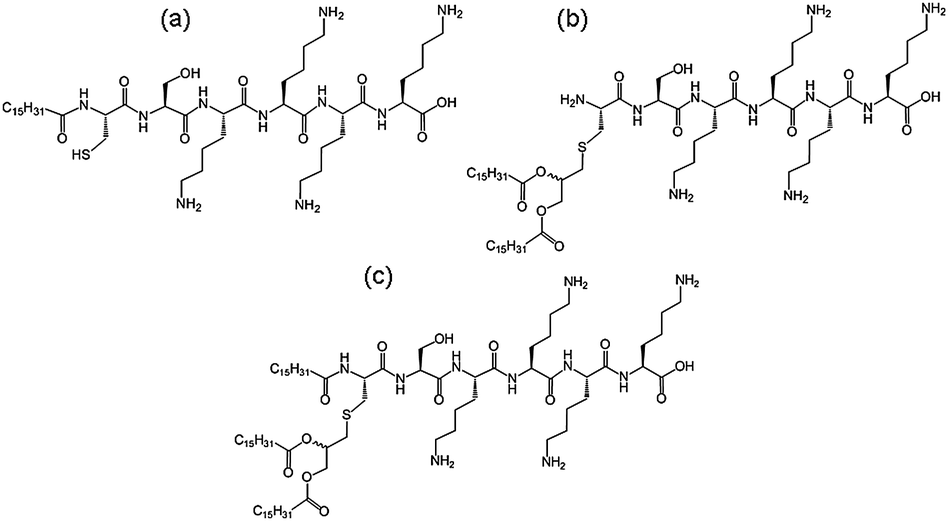 | ||
| Fig. 13 Structure of PamnCSK4 lipopeptides. (a) PamCSK4, (b) Pam2CSK4, (c) Pam3CSK4.71 | ||
 | ||
| Fig. 14 Cryo-TEM images obtained for 2 wt% solutions. (a) PamCSK, (b) Pam2CSK4, (c) Pam3CSK4. Enlarged images are shown at top in (a) and (b) and to the right in (c).71 | ||
The French National Agency for AIDS research (ANRS) has developed a series of HIV-lipopeptides as part of a strategy to create vaccines based on an induced CTL response.68 A list of typical structures is provided in Table 1. This builds on the use of simian immunodeficiency virus (SIV) as a model for vaccination using related lipopeptides (based on peptide sequences from the SIV NEF or GAG proteins) that induce a CTL response.75 To our knowledge, the self-assembly properties of these compounds has yet to be investigated.
| Lipo-peptide | Long peptide | Sequence | M W | No. AA |
|---|---|---|---|---|
| L-N1 | N1 (a.a. NEF66–97) | VGFPVTPQVPLRPMTYKAAVDLSHFLKEKGGLK(Pam) | 3862.8 | 32(+1) |
| L-N2 | N2 (a.a. NEF 117–147) | TQGYFPDWQNYTPGPGVRYPLTFGWCYKLVPK(Pam) | 4017.8 | 31(+1) |
| L-N3 | N3 (a.a. NEF182–205) | EWRFDSRLAFHHVARELHPEYFKNK(Pam) | 3451.1 | 24(+1) |
| L-G1 | G1 (a.a. GAG183–214) | DLNTMLNTVGGHQAAMQMLKETINEEAAEWDRK(Pam) | 3983.7 | 32(+1) |
| L-G2 | G2 (a.a. GAG253–284) | NPPIPVGEIYKRWIILGLNKIVRMYSPTSILDK(Pam) | 4063.1 | 32(+1) |
| L-E | E (a.a. ENV303–335) | TRPNNNTRKSIHIGPGRAFYATGEIIGDIRQAHK(Pam) | 4027.7 | 33(+1) |
A lipopeptide (termed “Theradigm”) used to create a vaccine against chronic hepatitis B virus (HBV) comprises a peptide sequence from a CTL linked to a T-helper peptide epitope TetTox and two palmitoyl lipid chains (Fig. 15).69 CTLs are able to recognise small 8–10 residue antigenic peptides. To our knowledge, the self-assembly behaviour of this lipopeptide has not yet been examined.
 | ||
| Fig. 15 Structure of theradigm lipopeptide.69 | ||
A lipopeptide with activity against HPV is C16-KSSTLGIVCPI which contains the TLGIVCPI peptide sequence associated with the E7 gene of HPV-16 as well as a hydrophilic KSS linker.70 The lipopeptide was shown to have activity in generating cellular immune responses in women with cervical cancer. A lipopeptide-based vaccine treatment for hepatitis C has also been trialled based on peptide sequences conjugated to the Pam2Cys lipopeptide.76
Lipopeptides for applications in cosmetics
We recently showed that the commercially available lipopeptide Matrixyl™ (C16-KTTKS) undergoes self-assembly in aqueous solution. This lipopeptide is used in skincare applications as an antiwrinkle activity is suggested.4,77 The peptide headgroup KTTKS is a sequence taken from a study on minimal Pro-collagen peptide sequences able to stimulate collagen and fibronectin production in fibroblasts.78At neutral pH and below the lipid chain melting temperature, highly extended nanotape structures are observed, as shown in Fig. 16.
 | ||
| Fig. 16 Fibrillar superstructure of peptide amphiphile Matrixyl.72 (a) Confocal optical microscopy image (fluorescent labelling with rhodamine B, 0.014 wt% Matrixyl in water), (b) apple green birefringence observed by polarized optical microscopy upon staining with Congo red, (c) differential optical microscopy image (1 mg ml−1 = 0.1 wt%). | ||
We showed that the bilayer nanotape self-assembled structure of C16-KTTKS is stable in the pH range 3–7 at room temperature, although there was evidence for twisting of the tapes at pH 4, but not pH 3 or pH 7.79 However, reduction of pH (to pH 2)79 or increase of temperature (above the lipid chain melting temperature, around 35 °C dependent on concentration)80 leads to the formation of spherical micelles. This is accompanied by a loss of β-sheet secondary structure, as revealed by circular dichroism spectroscopy and X-ray diffraction.79 Although very low pH is not relevant to the application of the compound in skincare (the pH of skin is around 4–6 although diseased skin can have lower pH), the lipid chain melting temperature is close to core body temperature although again the bilayer structure is expected to be stable near the surface of the skin in the stratum corneum.
This lipopeptide is able to stimulate collagen production by fibroblasts (dermal and corneal) in a dose-dependent manner close to the measured in DMEM media cac = 0.0055 wt%, as shown by the results from a Sirius red assay for type I collagen shown in Fig. 17.81 Since C16-KTTKS causes a slight decrease in cell number following initial cell seeding, the collagen production per cell increases more than the total collagen (Fig. 17). This study was the first systematic in vitro study on the collagen-stimulating properties of C16-KTTKS in the peer-reviewed literature.
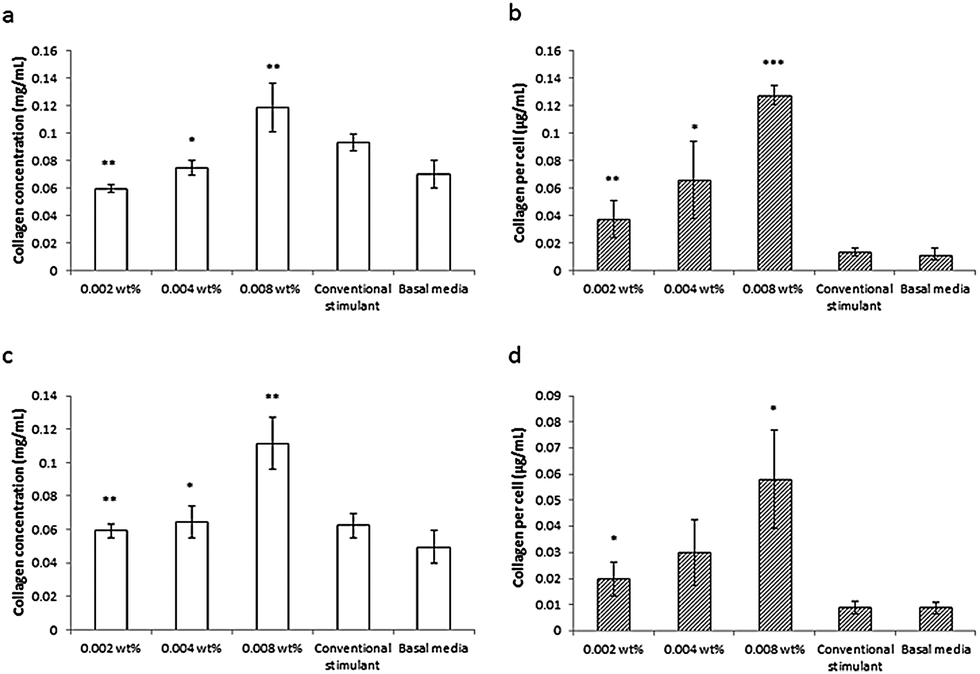 | ||
| Fig. 17 Collagen assay results showing the stimulation of human fibroblasts by C16-KTTKS.81 (a) and (b) Human corneal fibroblasts, (c) and (d) human dermal fibroblasts. Left, total amount of collagen deposited by cells and right, total amount of collagen produced per cell (n = 3, *P ≤ 0.005, **P ≤ 0.01 and ***P ≤ 0.001). | ||
We also examined the self-assembly in water of related lipopeptides of interest in skincare applications, C16-GHK and C16-KT as well as a commercial sample of C16-KTTKS (with higher polydispersity than the 95%+ purity grade sample used in our earlier studies).82 The KT peptide is a fragment of KTTKS while GHK is a copper-chelating peptide with a collagen-stimulating activity, involved in wound healing.83 All three lipopeptides showed similar cac values (although the transition is not as sharp as that observed for higher purity lipopeptides) and also had the common feature in SAXS of lamellar reflections indicating bilayer-based aggregates. However, TEM showed that C16-GHK and C16-KT form small crystallite structures whereas C16-KTTKS forms extended nanotapes. This latter material exhibits other “amyloid-like” aggregate features, including the uptake of the dye Congo red and a cross-β fibre X-ray diffraction pattern.82
Summary and future directions
This review has considered the bioactivity and self-assembly of three classes of lipopeptide (i) bacterially-expressed cyclic lipopeptides with major applications in practice as antibacterial and antifungal agents, (ii) linear toll-like receptor agonist lipopeptides with activity in immune therapies and (iii) several linear bio-derived lipopeptides with applications in skincare, in particular one with a collagen-stimulating activity (tradename Matrixyl™). In classes (ii) and (iii) recent work using cryo-TEM and SAXS has established the self-assembled nanostructure which is micelles or bilayer tapes, depending on the nature of the lipopeptide (in particular the number of lipid chains for the PamnCSK4 lipopeptides) as well as the self-assembly conditions (temperature, pH etc.). There are some studies on self-assembly of class (i) bacterially expressed lipopeptides with a cyclic peptide headgroup as cited above, but in many cases this has not yet been examined. Clearly, this is a fascinating direction for future research.One key question concerns the relationship, if any, between self-assembly and bioactivity. This is a complicated question for a number of reasons. Firstly, self-assembly is often concentration-dependent and decoupling the effect of concentration on bioactivity from the self-assembly process may be difficult. This may be circumvented by, for example, cross-linking self-assembled structures to trap a particular thermodynamic state. This itself may not be straightforward as the cross-linking process will influence the self-assembled structure and potentially have an impact on bioactivity. Second, it is not in general straightforward to probe interactions between the substrate and molecules and/or aggregate without perturbing the molecule-aggregate equilibrium, i.e. the self-assembly process, which is expected to be a closed process. There are likely to be cases where self-assembled structures themselves impart bioactivity – this is implied in much work on synthetic lipopeptides where lipopeptide assemblies present bioactive motifs at high density.84 This is also expected to be a significant effect for Matrixyl™ where the extended fibrils may provide a scaffold to enhance the collagen-stimulating activity of the peptide and/or provide a “filler” for anti-wrinkle skincare applications. It also seems likely to be the case that the aggregation of bacterially-expressed lipopeptides is important in stimulating or overcoming a host immune response. In other cases there may be no direct relationship between self-assembly and bioactivity (this will certainly be the case where bioactivity is observed at very low concentration below any cmc or cac). There may just be an indirect relationship in the sense that amphiphilicity is important to bioactivity (e.g. compatibility with the cell membrane) and the fact that this amphiphilicity in turn leads to self-assembly at high concentration. In cases such as the TLR agonist lipopeptides, crystal structures suggest binding sites for unaggregated molecules and the role of aggregate structures (if any) is not clear. The presence of possible oligomeric species on or off the aggregation pathway is a further complicating factor.
Moving forward, an interesting question is whether any general rules of self-assembly can be established for the large class of bacterially expressed lipopeptide, relating properties of the molecule (charge, shape) to aggregation propensity, self-assembled structure etc. In contrast to lipids, where models such as the surfactant packing parameter or interfacial geometry can be used to give guidance on the formation of self-assembled structures for a given molecule at a particular concentration,85 such models have yet to be developed for lipopeptides. This in part may reflect the complexity of intermolecular interactions involving both lipid chains and peptide headgroups. In the latter case, electrostatic, aromatic π-stacking and hydrogen bonding interactions are of competing importance depending on the sequence and this is counterbalanced by hydrophobic interactions of the lipid chains.
Acknowledgements
This work was supported by EPSRC grant EP/L020599/1. I am grateful to all collaborators who have worked in my group on some of the studies discussed herein including Dr Paula Jauregi (University of Reading) for the collaboration on B. subtilis lipopeptides, Prof. Janne Ruokolainen and his group (Aalto University) for the ongoing collaboration using cryo-TEM and to Dr Adam Round (ESRF) for help with BioSAXS at the ESRF, Grenoble, France.Notes and references
- (a) D. W. P. M. Löwik, E. H. P. Leunissen, M. van den Heuvel, M. B. Hansen and J. C. M. van Hest, Chem. Soc. Rev., 2010, 39, 3394–3412 RSC; (b) D. W. P. M. Löwik and J. C. M. van Hest, Chem. Soc. Rev., 2004, 33, 234–245 RSC; (c) H. G. Cui, M. J. Webber and S. I. Stupp, Pept. Sci., 2010, 94, 1–18 CrossRef CAS PubMed; (d) J. B. Matson, R. H. Zha and S. I. Stupp, Curr. Opin. Solid State Mater. Sci., 2011, 15, 225–235 CrossRef CAS PubMed; (e) J. Boekhoven and S. I. Stupp, Adv. Mater., 2014, 26, 1642–1659 CrossRef CAS PubMed; (f) I. W. Hamley, Soft Matter, 2011, 7, 4122–4138 RSC; (g) A. Dehsorkhi, V. Castelletto and I. W. Hamley, J. Pept. Sci., 2014, 20, 453–467 CrossRef CAS PubMed.
- (a) V. G. Fowler, Jr., H. W. Boucher, G. R. Corey, E. Abrutyn, A. W. Karchmer, M. E. Rupp, D. P. Levine, H. F. Chambers, F. P. Tally, G. A. Vigliani, C. H. Cabell, A. S. Link, I. DeMeyer, S. G. Filler, M. Zervos, P. Cook, J. Parsonnet, J. M. Bernstein, C. S. Price, G. N. Forrest, G. Faetkenheuer, M. Gareca, S. J. Rehm, H. R. Brodt, A. Tice, S. E. Cosgrove and S. A. E. Bacteremia, N. Engl. J. Med., 2006, 355, 653–665 CrossRef PubMed; (b) T. Owen, R. Pynn, J. S. Martinez and A. Butler, Langmuir, 2005, 21, 12109–12114 CrossRef CAS PubMed; (c) J. Hill, I. Parr, M. Morytko, J. Siedlecki, X. Y. Yu, J. Silverman, D. Keith, J. Finn, D. Christensen, T. Lazarova, A. D. Watson and Y. Zhang, Lipopeptides as antibacterial agents, U. S. Pat., US6911525B2, 2005 Search PubMed.
- (a) T. H. Wright, A. E. S. Brooks, A. J. Didsbury, G. M. Williams, P. W. R. Harris, P. R. Dunbar and M. A. Brimble, Angew. Chem., Int. Ed., 2013, 52, 10616–10619 CrossRef CAS PubMed; (b) W. G. Zeng, E. Eriksson, B. Chua, L. Grollo and D. C. Jackson, Amino Acids, 2010, 39, 471–480 CrossRef CAS PubMed.
- (a) K. Lintner and O. Peschard, Int. J. Cosmet. Sci., 2000, 22, 207–218 CrossRef CAS PubMed; (b) L. R. Robinson, N. C. Fitzgerald, D. G. Doughty, N. C. Dawes, C. A. Berge and D. L. Bissett, Int. J. Cosmet. Sci., 2005, 27, 155–160 CrossRef CAS PubMed.
- (a) J. M. Bonmatin, O. Laprevote and F. Peypoux, Comb. Chem. High Throughput Screening, 2003, 6, 541–556 CrossRef CAS; (b) P. Jacques, in Surfactin and other lipopeptides from Bacillus spp., ed. G. Soberon-Chavez, Springer, Berlin, 2011 Search PubMed.
- (a) M. Ongena and P. Jacques, Trends Microbiol., 2008, 16, 115–125 CrossRef CAS PubMed; (b) J. M. Raaijmakers, I. de Bruijn, O. Nybroe and M. Ongena, FEMS Microbiol. Rev., 2010, 34, 1037–1062 CrossRef CAS PubMed; (c) P. Jauregi, F. Coutte, L. Catiau, D. Lecouturier and P. Jacques, Sep. Purif. Technol., 2013, 104, 175–182 CrossRef CAS.
- T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020–13041 CrossRef CAS PubMed.
- H. Saito, S. Lund-Katz and M. C. Phillips, Prog. Lipid Res., 2004, 43, 350–380 CrossRef CAS PubMed.
- (a) E. Z. Ron and E. Rosenberg, Environ. Microbiol., 2001, 3, 229–236 CrossRef CAS PubMed; (b) P. Singh and S. S. Cameotra, Trends Biotechnol., 2004, 22, 142–146 CrossRef CAS PubMed.
- (a) R. Maget-Dana and F. Peypoux, Toxicology, 1994, 87, 151–174 CrossRef CAS PubMed; (b) F. Peypoux, J. M. Bonmatin and J. Wallach, Appl. Microbiol. Biotechnol., 1999, 51, 553–563 CrossRef CAS PubMed; (c) G. Seydlova and J. Svobodova, Cent. Eur. J. Med., 2008, 3, 123–133 CAS; (d) I. M. Banat, A. Franzetti, I. Gandolfi, G. Bestetti, M. G. Martinotti, L. Fracchia, T. J. Smyth and R. Marchant, Appl. Microbiol. Biotechnol., 2010, 87, 427–444 CrossRef CAS PubMed; (e) J. M. Raaijmakers, I. de Bruijn and M. J. D. de Kock, Mol. Plant-Microbe Interact., 2006, 19, 699–710 CrossRef CAS PubMed.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef CAS PubMed.
- (a) V. Miao, M. F. Coeffet-LeGal, P. Brian, R. Brost, J. Penn, A. Whiting, S. Martin, R. Ford, I. Parr, M. Bouchard, C. J. Silva, S. K. Wrigley and R. H. Baltz, Microbiology, 2005, 151, 1507–1523 CrossRef CAS PubMed; (b) J. N. Steenbergen, J. Alder, G. M. Thorne and F. P. Tally, J. Antimicrob. Chemother., 2005, 55, 283–288 CrossRef CAS PubMed.
- M. Debono, B. J. Abbott, R. M. Molloy, D. S. Fukuda, A. H. Hunt, V. M. Daupert, F. T. Counter, J. L. Ott, C. B. Carrell, L. C. Howard, L. D. Boeck and R. L. Hamill, J. Antibiot., 1988, 41, 1093–1105 CrossRef CAS PubMed.
- (a) J. K. Muraih and M. Palmer, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 1642–1647 CrossRef CAS PubMed; (b) J. Qiu and L. E. Kirsch, J. Pharm. Sci., 2014, 103, 853–861 CrossRef CAS PubMed.
- J. A. Silverman, N. G. Perlmutter and H. M. Shapiro, Antimicrob. Agents Chemother., 2003, 47, 2538–2544 CrossRef CAS PubMed.
- T. H. Zhang, J. K. Muraih, B. MacCormick, J. Silverman and M. Palmer, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 2425–2430 CrossRef CAS PubMed.
- (a) J. H. Lakey and M. Ptak, Biochemistry, 1988, 27, 4639–4645 CrossRef CAS PubMed; (b) D. Jung, A. Rozek, M. Okon and R. E. W. Hancock, Chem. Biol., 2004, 11, 949–957 CrossRef CAS PubMed; (c) D. Jung, J. P. Powers, S. K. Straus and R. E. W. Hancock, Chem. Phys. Lipids, 2008, 154, 120–128 CrossRef CAS PubMed.
- I. W. Hamley, S. Kirkham, M. Reza and J. Ruokolainen, 2015, unpublished work.
- D. W. Denning, Lancet, 2003, 362, 1142–1151 CrossRef CAS.
- M. Debono and R. S. Gordee, Annu. Rev. Microbiol., 1994, 48, 471–497 CrossRef CAS PubMed.
- (a) N. Milton, K. P. Moder, J. L. Sabatowski and S. A. Sweetana, US Pat., US06960564, 2005 Search PubMed; (b) N. Milton, K. P. Moder, J. L. Sabatowski and S. A. Sweetana, US Pat., US07709444, 2010 Search PubMed.
- M. Mazzola, I. de Bruijn, M. F. Cohen and J. M. Raaijmakers, Appl. Environ. Microbiol., 2009, 75, 6804–6811 CrossRef CAS PubMed.
- (a) T. R. Neu, T. Hartner and K. Poralla, Appl. Microbiol. Biotechnol., 1990, 32, 518–520 CAS; (b) H. S. Saini, B. E. Barragan-Huerta, A. Lebron-Paler, J. E. Pemberton, R. R. Vazquez, A. M. Burns, M. T. Marron, C. J. Seliga, A. A. L. Gunatilaka and R. M. Maier, J. Nat. Prod., 2008, 71, 1011–1015 CrossRef CAS PubMed.
- V. Groupe, L. H. Pugh, D. Weiss and M. Kochi, Proc. Soc. Exp. Biol. Med., 1951, 78, 354–358 CrossRef CAS PubMed.
- S. Lang, Curr. Opin. Colloid Interface Sci., 2002, 7, 12–20 CrossRef CAS.
- (a) http://www.lipidmaps.org/data/LMSDRecord.php?Mode=View&ViewType=ChemDraw&LMID=LMPK14000008, in 2015; (b) A. Kwa, S. K. Kosiakou, V. H. Tam and M. E. Falagas, Expert Rev. Anti-Infect. Ther., 2007, 5, 811–821 CrossRef CAS PubMed.
- (a) M. M. Yakimov, K. N. Timmis, V. Wray and H. L. Fredrickson, Appl. Environ. Microbiol., 1995, 61, 1706–1713 CAS; (b) I. Grangemard, J. Wallach, R. Maget-Dana and F. Peypoux, Appl. Biochem. Biotechnol., 2001, 90, 199–210 CrossRef CAS PubMed.
- N. Naruse, O. Tenmyo, S. Kobaru, H. Kamei, T. Miyaki, M. Konishi and T. Oki, J. Antibiot., 1990, 43, 267–280 CrossRef CAS PubMed.
- D. Landman, C. Georgescu, D. A. Martin and J. Quale, Clin. Microbiol. Rev., 2008, 21, 449–465 CrossRef CAS PubMed.
- I. W. Hamley, A. Dehsorkhi, P. Jauregi, J. Seitsonen, J. Ruokolainen, F. Coutte, G. Chataigné and P. Jacques, Soft Matter, 2013, 9, 9572–9578 RSC.
- M. Ongena, P. Jacques, Y. Toure, J. Destain, A. Jabrane and P. Thonart, Appl. Microbiol. Biotechnol., 2005, 69, 29–38 CrossRef CAS PubMed.
- D. Julkowska, M. Obuchowski, I. B. Holland and S. J. Séror, J. Bacteriol., 2005, 187, 65–76 CrossRef CAS PubMed.
- W. Li, H. Rokni-Zadeh, M. De Vleeschouwer, M. G. K. Ghequire, D. Sinnaeve, G. L. Xie, J. Rozenski, A. Madder, J. C. Martins and R. De Mot, PLoS One, 2013, 8, e62946 CAS.
- M. L. Hutchison and D. C. Gross, Mol. Plant-Microbe Interact., 1997, 10, 347–354 CrossRef CAS PubMed.
- D. Sorensen, T. H. Nielsen, C. Christophersen, J. Sorensen and M. Gajhede, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 1123–1124 CAS.
- D. P. Galonic, E. W. Barr, C. T. Walsh, J. M. Bollinger and C. Krebs, Nat. Chem. Biol., 2007, 3, 113–116 CrossRef CAS PubMed.
- A. Andolfi, A. Cimmino, P. L. Cantore, N. S. Iacobellis and A. Evidente, Perspect. Med. Chem., 2008, 2, 81–112 CAS.
- H. Heerklotz and J. Seelig, Biophys. J., 2001, 81, 1547–1554 CrossRef CAS PubMed.
- H. H. Shen, R. K. Thomas, C. Y. Chen, R. C. Darton, S. C. Baker and J. Penfold, Langmuir, 2009, 25, 4211–4218 CrossRef CAS PubMed.
- H. H. Shen, R. K. Thomas and P. Taylor, Langmuir, 2010, 26, 320–327 CrossRef CAS PubMed.
- Y. Ishigami, M. Osman, H. Nakahara, Y. Sano, R. Ishiguro and M. Matsumoto, Colloids Surf., B, 1995, 4, 341–348 CrossRef CAS.
- I. Harnois, D. Genest, J. C. Brochon and M. Ptak, Biopolymers, 1988, 27, 1403–1413 CrossRef CAS PubMed.
- A. Grau, J. C. Gomez-Fernandez, F. Peypoux and A. Ortiz, Peptides, 2001, 22, 1–5 CrossRef CAS.
- R. Maget-Dana, M. Ptak, F. Peypoux and G. Michel, Biochim. Biophys. Acta, 1985, 815, 405–409 CrossRef CAS.
- C. Carrillo, J. A. Teruel, F. J. Aranda and A. Ortiz, Biochim. Biophys. Acta, Biomembr., 2003, 1611, 91–97 CrossRef CAS.
- J. D. Sheppard, C. Jumarie, D. G. Cooper and R. Laprade, Biochim. Biophys. Acta, 1991, 1064, 13–23 CrossRef CAS.
- H. Heerklotz and J. Seelig, Eur. Biophys. J. Biophys. Lett., 2007, 36, 305–314 CrossRef CAS PubMed.
- D. Vollenbroich, M. Ozel, J. Vater, R. M. Kamp and G. Pauli, Biologicals, 1997, 25, 289–297 CrossRef CAS PubMed.
- K. Kim, S. Y. Jung, D. K. Lee, J. K. Jung, J. K. Park, D. K. Kim and C. H. Lee, Biochem. Pharmacol., 1998, 55, 975–985 CrossRef CAS PubMed.
- J. R. Mireles, A. Toguchi and R. M. Harshey, J. Bacteriol., 2001, 183, 5848–5854 CrossRef CAS PubMed.
- T. Kikuchi and K. Hasumi, Biochim. Biophys. Acta, 2002, 1596, 234–245 CrossRef CAS.
- S. Dufour, M. Deleu, K. Nott, B. Wathelet, P. Thonart and M. Paquot, Biochim. Biophys. Acta, Gen. Subj., 2005, 1726, 87–95 CrossRef CAS PubMed.
- N. J. Gay and M. Gangloff, Annu. Rev. Biochem., 2007, 76, 141–165 CrossRef CAS PubMed.
- E. J. Mifsud, A. C. L. Tan and D. C. Jackson, Front. Immunol., 2014, 5, 1–10 CAS.
- J. Y. Kang, X. Nan, M. S. Jin, S. J. Youn, Y. H. Ryu, S. Mah, S. H. Han, H. Lee, S. G. Paik and J. O. Lee, Immunity, 2009, 31, 873–884 CrossRef CAS PubMed.
- M. S. Jin, S. E. Kim, J. Y. Heo, M. E. Lee, H. M. Kim, S. G. Paik, H. Y. Lee and J. O. Lee, Cell, 2007, 130, 1071–1082 CrossRef CAS PubMed.
- V. Braun, Biochim. Biophys. Acta, 1975, 415, 335–377 CrossRef CAS.
- W. G. Zeng, S. Ghosh, Y. F. Lau, L. E. Brown and D. C. Jackson, J. Immunol., 2002, 169, 4905–4912 CrossRef.
- K. Deres, H. Schild, K. H. Wiesmuller, G. Jung and H. G. Rammensee, Nature, 1989, 342, 561–564 CrossRef CAS PubMed.
- K. H. Wiesmuller, G. Jung and G. Hess, Vaccine, 1989, 7, 29–33 CrossRef CAS PubMed.
- (a) P. F. Muhlradt, M. Kiess, H. Meyer, R. Sussmuth and G. Jung, J. Exp. Med., 1997, 185, 1951–1958 CrossRef CAS PubMed; (b) P. F. Muhlradt, M. Kiess, H. Meyer, R. Sussmuth and G. Jung, Infect. Immun., 1998, 66, 4804–4810 CAS.
- M. Morr, O. Takechi, S. Akira, M. M. Simon and P. F. Mühlradt, Eur. J. Immunol., 2002, 32, 3337–3347 CrossRef CAS PubMed.
- O. Takeuchi, T. Kawai, P. F. Muhlradt, M. Morr, J. D. Radolf, A. Zychlinsky, K. Takeda and S. Akira, Int. Immunol., 2001, 13, 933–940 CrossRef CAS PubMed.
- (a) O. Takeuchi, S. Sato, T. Horiuchi, K. Hoshino, K. Takeda, Z. Y. Dong, R. L. Modlin and S. Akira, J. Immunol., 2002, 169, 10–14 CrossRef CAS PubMed; (b) K. O. Omueti, J. M. Beyer, C. M. Johnson, E. A. Lyle and R. I. Tapping, J. Biol. Chem., 2005, 280, 36616–36625 CrossRef CAS PubMed.
- A. C. L. Tan, E. J. Mifsud, W. G. Zeng, K. Edenborough, J. McVernon, L. E. Brown and D. C. Jackson, Mol. Pharmaceutics, 2012, 9, 2710–2718 CrossRef CAS PubMed.
- B. Y. Chua, D. Pejoski, S. J. Turner, W. G. Zeng and D. C. Jackson, J. Immunol., 2011, 187, 1692–1701 CrossRef CAS PubMed.
- T. H. Wright, A. E. S. Brooks, A. J. Didsbury, G. M. Williams, P. W. R. Harris, P. R. Dunbar and M. A. Brimble, Angew. Chem., Int. Ed., 2013, 52, 10616–10619 CrossRef CAS PubMed.
- (a) G. Pialoux, H. Gahery-Segard, S. Sermet, H. Poncelet, S. Fournier, L. Gerard, A. Tartar, H. Gras-Masse, J. P. Levy, J. G. Guillet and A. V. S. Team, AIDS, 2001, 15, 1239–1249 CrossRef CAS PubMed; (b) C. Durier, O. Launay, V. Meiffredy, Y. Saidi, D. Salmon, Y. Levy, J. G. Guillet, G. Pialoux and J. P. Aboulker, AIDS, 2006, 20, 1039–1049 CrossRef CAS PubMed.
- A. Vitiello, G. Ishioka, H. M. Grey, R. Rose, P. Farness, R. Lafond, L. L. Yuan, F. V. Chisari, J. Furze, R. Bartholomeuz and R. W. Chesnut, J. Clin. Invest., 1995, 95, 341–349 CrossRef CAS PubMed.
- M. A. Steller, K. J. Gurski, M. Murakami, R. W. Daniel, K. V. Shah, E. Celis, A. Sette, E. L. Trimble, R. C. Park and F. M. Marincola, Clin. Cancer Res., 1998, 4, 2103–2109 CAS.
- I. W. Hamley, S. Kirkham, A. Dehsorkhi, V. Castelletto, M. Reza and J. Ruokolainen, Chem. Commun., 2014, 50, 15948–15951 RSC.
- V. Castelletto, I. W. Hamley, J. Perez, L. Abezgauz and D. Danino, Chem. Commun., 2010, 46, 9185–9187 RSC.
- A. Brizard, C. Aime, T. Labrot, I. Huc, D. Berthier, F. Artzner, B. Desbat and R. Oda, J. Am. Chem. Soc., 2007, 129, 3754–3762 CrossRef CAS PubMed.
- A. B. Schromm, J. Howe, A. J. Ulmer, K. H. Wiesmuller, T. Seyberth, G. Jung, M. Rossle, M. H. J. Koch, T. Gutsmann and K. Brandenburg, J. Biol. Chem., 2007, 282, 11030–11037 CrossRef CAS PubMed.
- I. Bourgault, F. Chirat, A. Tartar, J. P. Levy, J. G. Guillet and A. Venet, J. Immunol., 1994, 152, 2530–2537 CAS.
- E. M. Y. Eriksson and D. C. Jackson, Curr. Protein Pept. Sci., 2007, 8, 412–417 CrossRef CAS PubMed.
- K. Lintner, US Pat., US2004/0132667, 2003 Search PubMed.
- K. Katayama, J. Armendarizborunda, R. Raghow, A. H. Kang and J. M. Seyer, J. Biol. Chem., 1993, 268, 9941–9944 CAS.
- A. Dehsorkhi, V. Castelletto, I. W. Hamley, J. Adamcik and R. Mezzenga, Soft Matter, 2013, 9, 6033–6036 RSC.
- J. F. Miravet, B. Escuder, M. D. Segarra-Maset, M. Tena-Solsona, I. W. Hamley, A. Dehsorkhi and V. Castelletto, Soft Matter, 2013, 9, 3558–3564 RSC.
- R. R. Jones, V. Castelletto, C. J. Connon and I. W. Hamley, Mol. Pharmaceutics, 2013, 10, 1063–1069 CrossRef CAS PubMed.
- V. Castelletto, I. W. Hamley, C. Whitehouse, P. Matts, R. Osborne and E. S. Baker, Langmuir, 2013, 29, 9149–9155 CrossRef CAS PubMed.
- (a) F. X. Maquart, L. Pickart, M. Laurent, P. Gillery, J. C. Monboisse and J. P. Borel, FEBS Lett., 1988, 238, 343–346 CrossRef CAS PubMed; (b) F. X. Maquart, G. Bellon, B. Chaqour, J. Wegrowski, L. M. Patt, R. E. Trachy, J. C. Monboisse, F. Chastang, P. Birembaut, P. Gillery and J. P. Borel, J. Clin. Invest., 1993, 92, 2368–2376 CrossRef CAS PubMed.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2012, 48, 26–33 RSC.
- I. W. Hamley, Introduction to Soft Matter, Wiley, revised edn, 2007 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |