
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An extended framework of cages formed of pre-synthesised and functionalised heterometallic cages†
George F. S.
Whitehead
a,
Simon J.
Teat
b,
Kevin J.
Gagnon
b,
Grigore A.
Timco
a and
Richard E. P.
Winpenny
*a
aSchool of Chemistry, The University of Manchester, Oxford Road, M13 9PL, Manchester, UK. E-mail: richard.winpenny@manchester.ac.uk; Fax: +44 (0)161-275-4616
bAdvanced Light Source, Lawrence Berkeley Laboratory, 1 Cyclotron Road, MS2-400, Berkeley, California 94720, USA
First published on 3rd February 2015
Abstract
We present the first example of an extended 3D framework synthesised without the use of an isolable organic component. This is achieved by the combination of two cage complexes isolated and functionalised prior to synthesis; [nPr2NH2][Cr7NiF8(O2CtBu)14(O2CC5H4N)2] and [Fe2CoO(O2CtBu)6(HO2CtBu)3], where the former is as a bidentate linker, bridging between three nodes of the latter.
The field of metal organic frameworks (MOFs) is one of the most actively pursued areas of chemistry,1 with potential applications of frameworks ranging from gas storage and separation, catalysis, optical and ferroelectric properties.2 Generally, they consist of either individual metal sites, or simple polymetallic clusters with multiple binding sites, linked by polydentate organic molecules. The wide array of metal salts, simple clusters and polydentate organic molecules has led to over 20
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 different MOFs being reported in the last decade.3 However, in all reported cases there is always an isolable organic component bridging between a metal-containing components. In some cases the metal containing component forms serendipitously during the reaction and is not isolable for characterisation prior to the formation of the framework. This can limit control over the nature of the cages within the framework and therefore the overall properties of the framework.
000 different MOFs being reported in the last decade.3 However, in all reported cases there is always an isolable organic component bridging between a metal-containing components. In some cases the metal containing component forms serendipitously during the reaction and is not isolable for characterisation prior to the formation of the framework. This can limit control over the nature of the cages within the framework and therefore the overall properties of the framework.
We propose a new route to synthesise such materials, avoiding a purely organic linker by using functionalised polymetallic cages, which by virtue of their functionality can act as a linker to other cages. This allows the formation of structures containing dissimilar polymetallic cages, where both have been pre-synthesised with desired properties to produce a hybrid material with retention of those properties. Here we present the preliminary work towards this goal with the structure of a new material synthesised by such methods.
The approach we have taken here is to introduce multiple Lewis base functional sites to the periphery of a pre-synthesised cage. This allows the functionalised cage to bind to Lewis acidic sites on other cages, thereby facilitating a link between the two. For our initial studies our Lewis base functionalised cage is a variant of [nPr2NH2][Cr7NiF8(O2CtBu)16], 1 a heterometallic cage consisting of a regular octagon of seven chromium(III) and one nickel(II), bridged on the inner edges by eight fluorides and the outer edge by sixteen pivalate ligands.4 The pivalates are arranged so that eight reside in the equatorial plane of the metal sites and the remaining eight reside axial to the metal plane, alternating above and below the plane. The whole assembly is anionic and at the centre is a central ammonium cation, which is thought to act as a template for the ring formation.
Lewis base functionality is introduced to 1 by substitution of the peripheral pivalate ligands with iso-nicotinic acid.5 This introduces outward facing pyridyl groups to the periphery of 1. Substitution occurs only on the Cr–Ni edges of the cage by virtue of the differences in kinetics of chromium(III) and nickel(II), of which the nickel(II) is expected to react approximately 1010 times faster.6 This is achieved by refluxing 1 in the presence of iso-nicotinic acid in n-propanol for 24 hours, producing [nPr2NH2][Cr7NiF8(O2CtBu)16−x(O2CC5H4N)x], where x = 1 and 2 (2 and 3, respectively). 2, a mono-axial substituted derivative, is the major product from the reaction and has been shown to act as a ligand towards single metal sites and polymetallic clusters producing large, 0D assemblies of clusters, or “cages-of-cages”.73, the axial–axial disubstituted derivative, is an easily isolated minor product that has been shown to act as a Lewis base towards metal sites to produce 1D polymer chains.5 Here we discuss the use of the 3 (Fig. 1), as a ligand in framework synthesis.
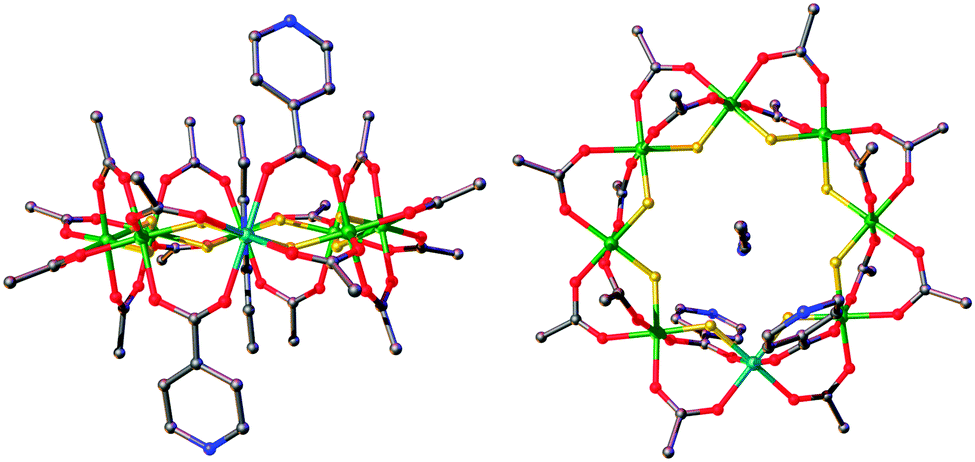 | ||
| Fig. 1 Structure of 3 in the crystal. Green = chromium, turquoise = nickel, grey = carbon, red = oxygen, blue = nitrogen, yellow = fluorine. | ||
3 can be regarded as a bidentate ligand, analogous to 4,4′-bipyridyl but with significant differences between the two. Firstly the presence of the {Cr7Ni} ring in 3 puts extreme steric demands on the ligand and significantly alters the solubility. Secondly, the pyridyl groups are not linearly disposed, due to the axial–axial substituted nature of the ring resulting in the iso-nicotinate groups residing on different edges of the ring. These are offset by 3.15 Å at an angle of 136.2°, close to the internal angle of a regular octagon.53 is also a chiral molecule, as dictated by the directions of the pyridyl groups on the edge of the ring, and forms as a racemate during synthesis.
A classic route to MOFs often involves a linear linker (here 3) with a three-connecting node. For example, {M2M′O(O2CR)6} triangles have been used extensively in the formation of porous framework materials, forming both 2D and 3D structures.8–10 These are generally made by solvothermal synthesis with the triangle being formed in situ by reaction of a metal salt with a either a pyridyl containing carboxylic acid or a multi-dentate carboxylic acid. A more controlled method of synthesis involves the pre-synthesis of Fe2M(μ3-O)(O2CtBu)6 heterometallic variants and reacting these with bidentate N-donor molecules. These have also been shown to form 2D honeycomb networks, both interpenetrating and stacked, and 3D interpenetrated frameworks with bidentate and tridentate linkers.11–13 There are three possible frameworks that could result from a linear bidentate linker and a three connected node assuming interpenetration of the network. These are 6,3 nets with varying degrees and morphologies of interpenetration, depending on if the framework formed is 2D or 3D; 10,3 nets with two different morphologies, 10,3a or 10,3b, or very rarely an 8,3c net.1a The preference for which net forms is related to the steric demands of components and/or control of chirality at the three-connecting points.
We have reacted 3 with a range of oxo-centred metal triangles, chosen as the simplest motif with enough steric freedom and sufficient binding sites to produce an extended structure. We find that with [Fe2Co(μ3-O)(O2CtBu)6(HO2CtBu)3], 4,14 the three labile terminal pivalate ligands can be displaced by the pyridyls of 3, with the reaction of the two in hot acetone initially forming an amorphous powder, insoluble in all solvents with the exception of THF. Characterisation of the resulting compound is challenging; repeated recrystallization of the powder by slow evaporation of THF was required to produce crystals suitable for single crystal diffraction studies. The results show the formation of the unprecedented extended framework structure 5 where both components are polymetallic cages (Fig. 2).
 | ||
| Fig. 2 (a) Orthographic view of 5 with one net coloured light blue to highlight interpenetrated nature. (b) Showing a single net. t-Butyl groups of pivalate groups and central ammonium cations removed for clarity. Green = chromium, turquoise = nickel, orange = iron/cobalt, grey = carbon, red = oxygen, blue = nitrogen, yellow = fluorine. | ||
The structure consists of 4 bridged by three 3 from the labile ligand sites, which in turn further bridge to another 4, forming an extended 3D network. Simplification and analysis of this network with TOPOS shows that it is a 10,3 net with 2-fold interpenetration and closer inspection revels it to be a 10,3b net.15 The arrangement is typical of 10,3b net;16 alternating layers of aligned 1D planar zig-zag chains of 3 bridging between 4, which in turn are cross-linked by extra 3 between the third nodes of 4, which are directed above and below of the chains. The 1D planar zigzag arrangement has a similar geometry to those seen previously with 1D chains synthesised with 3.5 Evidently, this is the most stable geometry for this class of compound, driven by the steric demands of 3.
As there is a third binding site on each {Fe2Co} triangle further crosslinking by 3 is possible. This results in crosslinking between the neighbouring chains and establishes the 10,3b net framework structure (Fig. 2). The framework is distorted away from the perfect tetragonal 10,3b form, most likely due to the very bulky {Cr7Ni} ring, and the alternating layers propagate at an angle of 63° to each other. The arrangement is complemented by an interpenetrating counterpart.
Chiral selectivity of 3 within the framework is needed in order to maintain the symmetry of the MOF. In the case of the 1D zig-zag motifs, the two enantiomers of 3 alternate along the length of the polymer chain to maintain the linearity of the chain (Fig. 3). As for the crosslinking 3 between the 1D zigzag planes, these alternate in chirality for every interchain plane i.e., each layer of cross linkers between the planes of 1D chains is enantiomerically pure.
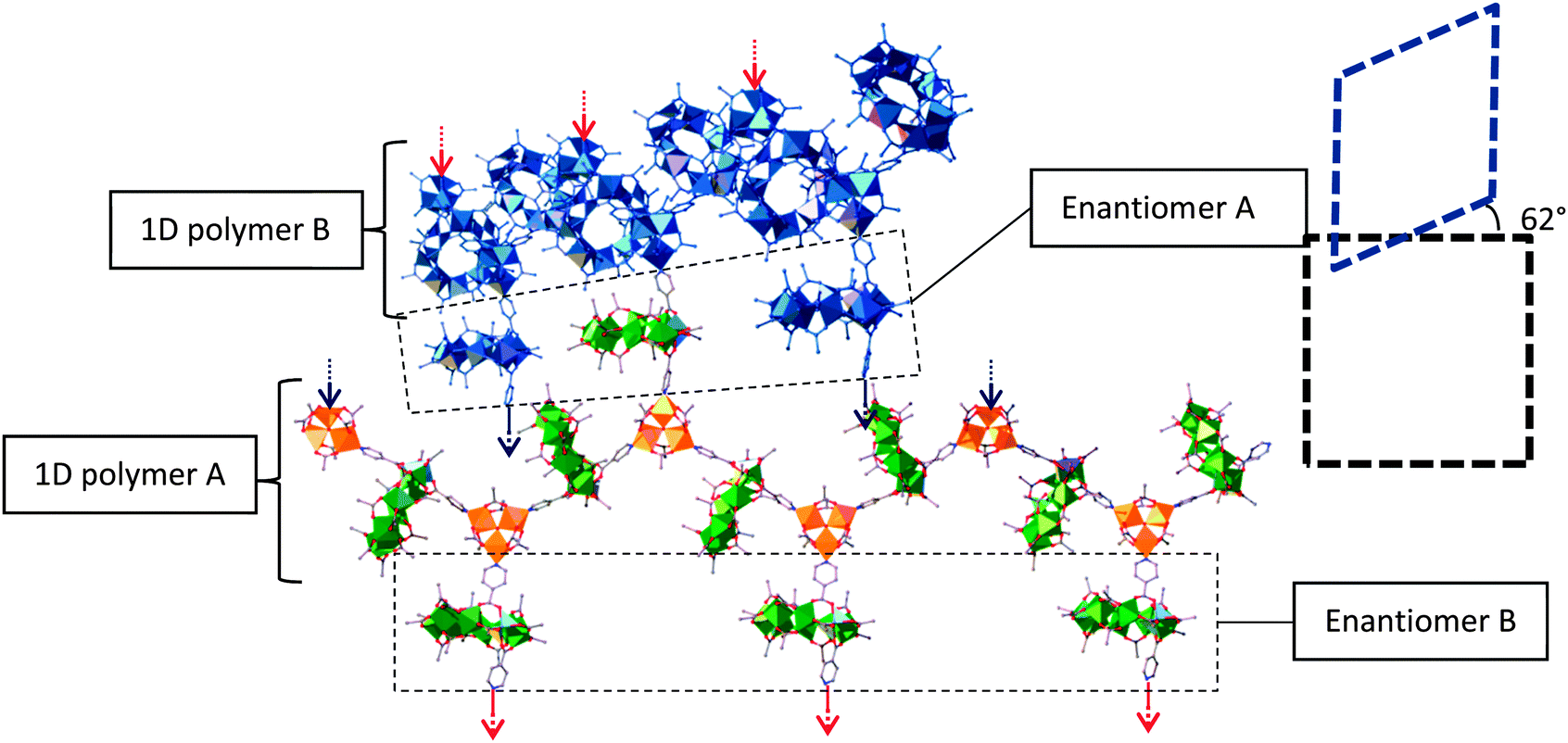 | ||
| Fig. 3 Showing the 1D chains and crosslinking within the framework. Colours as Fig. 2. Blue chain represents neighbouring polymer chain. The red and blue arrows represent where the crosslinks of each chain bind to the polymers in the neighbouring planes. The dashed boxes represent the angle of offset between the chains in the neighbouring planes. | ||
This demand for stereoselectivity could help explain the difficulties encountered during crystallisation of the framework, with very slow crystallisation needed to grow crystals of sufficient quality for X-ray single crystal diffraction. Fast crystallisation results in large blocks of a highly crystalline material. The same unit cell as that found for 5 can be determined for these crystals, but diffraction is limited to a resolution of approximately 4 Å, which is insufficient to solve the structure. This could imply some disorder between enantiomers of 3 within these crosslinking layers leading to diffuse scattering and a lack of short range order.
The mild conditions of the reaction rule out metal exchange between 3 and 4, so the metal sites reside where they would be expected to be. Within the {Cr7Ni} rings the positions of the two metals are fully-determined, as the Ni site is the vertex of the octagon that is bound to both iso-nicotinate ligands. Unfortunately, due to the symmetry of 4 the iron and cobalt sites within the triangles cannot be determined crystallographically and are assumed to be disordered equally over all three sites.
The interpenetrated structure has a large void volume of 81340 Å3, which is approximately 52% of the overall structure. This is occupied by disordered solvent molecules; the positions of these molecules could not be determined by X-ray diffraction, which suggests they are significantly disordered. Removing crystals of 5 from the mother liquor leads to rapid desolvation and loss of crystallinity; all attempts to prepare powder samples of 5 for diffraction studies led to amorphous materials due to desolvation. Similarly attempts to perform thermogravimetric analysis showed little detail as preparation of the sample led to immediate solvent loss. Thereafter the sample was stable until over 230°, after which the sample begins to decompose.†
As far as we can tell, this is the first example of an extended framework structure where preformed cages bridge to other preformed cages without the requirement of an added organic linker or the serendipitous assembly of cages in situ to complete the structure. Further investigation into the gas sorption properties of these complexes will be undertaken, as will the synthesis and characterisation of new frameworks. Overall, the ability to form these structures furthers progress towards our ultimate goal of controlled assembly of molecular nanomagnets into complex architectures.
This research was funded by the EPSRC (UK) through grants. R.E.P.W. thanks the Royal Society for a Wolfson Research Merit Award.
Notes and references
- (a) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef; (b) R. Robson, J. Chem. Soc., Dalton Trans., 2000, 3735 RSC; (c) A. J. Blake, N. R. Champness, P. Hubberstey, W. S. Li, M. A. Withersby and M. Schroder, Coord. Chem. Rev., 1999, 183, 117 CrossRef CAS; (d) O. M. Yaghi, C. E. Davis, G. M. Li and H. Li, J. Am. Chem. Soc., 1997, 119, 2861 CrossRef CAS; (e) M. J. Zaworotko, Angew. Chem., Int. Ed., 2000, 39, 3052 CrossRef CAS; (f) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed; (g) J. S. Seo, D. Wang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982 CrossRef CAS PubMed; (h) D. M. L. Goodgame, D. A. Grachvogel and D. J. Williams, Angew. Chem., Int. Ed., 1999, 38, 153 CrossRef CAS.
- Functional Metal-Organic Frameworks: Gas Storage, Separation and Catalysis, ed. M. Schroder, Springer-Verlag Berlin, Germany, 2010 Search PubMed.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhan, Q. Zhang, T. Gentle Iii, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561 RSC , and references therein.
- (a) F. K. Larsen, E. J. L. McInnes, H. El Mkami, J. Overgaard, S. Piligkos, G. Rajaraman, E. Rentschler, A. A. Smith, G. M. Smith, V. Boote, M. Jennings, G. A. Timco and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2003, 42, 101 CrossRef CAS; (b) G. A. Timco, T. B. Faust, F. Tuna and R. E. P. Winpenny, Chem. Soc. Rev., 2011, 40, 3067 RSC.
- G. F. S. Whitehead, B. Cross, L. Carthy, V. A. Milway, H. Rath, A. Fernandez, S. L. Heath, C. A. Muryn, R. G. Pritchard, S. J. Teat, G. A. Timco and R. E. P. Winpenny, Chem. Commun., 2013, 49, 7195 RSC.
- S. F. Lincoln, Helv. Chim. Acta, 2005, 88, 523 CrossRef CAS.
- G. F. S. Whitehead, F. Moro, G. A. Timco, W. Wernsdorfer, S. J. Teat and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2013, 52, 9932 CrossRef CAS PubMed.
- A. Schoedel and M. J. Zaworotko, Chem. Sci., 2014, 5, 1269 RSC.
- C. Serre, F. Millange, S. Surblé and G. Férey, Angew. Chem., Int. Ed., 2004, 43, 6285 CrossRef PubMed.
- K. Barthelet, D. Riou and G. Ferey, Chem. Commun., 2002, 1492 RSC.
- R. A. Polunin, S. V. Kolotilov, M. A. Kiskin, O. Cador, E. A. Mikhalyova, A. S. Lytvynenko, S. Golhen, L. Ouahab, V. I. Ovcharenko, I. L. Eremenko, V. M. Novotortsev and V. V. Pavlishchuk, Eur. J. Inorg. Chem., 2010, 5055 CrossRef CAS.
- R. A. Polunin, S. V. Kolotilov, M. A. Kiskin, O. Cador, S. Golhen, O. V. Shvets, L. Ouahab, Z. V. Dobrokhotova, V. I. Ovcharenko, I. L. Eremenko, V. M. Novotortsev and V. V. Pavlishchuk, Eur. J. Inorg. Chem., 2011, 4985 CrossRef CAS.
- O. Botezat, J. van Leusen, V. C. Kravtsov, I. G. Filippova, J. Hauser, M. Speldrich, R. P. Hermann, K. W. Krämer, S.-X. Liu, S. Decurtins, P. Kögerler and S. G. Baca, Cryst. Growth Des., 2014, 14, 4721 CAS.
- K. O. Abdulwahab, M. A. Malik, P. O'Brien, G. A. Timco, F. Tuna, C. A. Muryn, R. E. P. Winpenny, R. A. D. Pattrick, V. S. Coker and E. Arenholz, Chem. Mater., 2014, 4, 999–1013 CrossRef.
- V. A. Balatov, IUCr CompComm Newslett., 2006, 7, 4 Search PubMed.
- S. R. Batten, S. M. Neville and D. R. Turner, Coordination Polymers: Design, Analysis and Application, The Royal Society of Chemistry, Cambridge, 2009, ch. 2, p. 37 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of synthesis and structural studies. CCDC 1039416. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc10035b |
| This journal is © The Royal Society of Chemistry 2015 |