
Large-scale production of spherical Y2O3:Eu3+ phosphor powders with narrow size distribution using a two-step spray drying method†
Jung Sang Cho‡
a,
Kyeong Youl Jung‡b,
Mun Young Sonc and
Yun Chan Kang*a
aDepartment of Materials Science and Engineering, Korea University, Anam-Dong, Seongbuk-Gu, Seoul 136-713, Republic of Korea. E-mail: yckang@korea.ac.kr; Fax: +82-2-928-3584
bDepartment of Chemical Engineering, Kongju National University, 1223-24 Cheonan-Daero, Seobuk-gu, Cheonan, Republic of Korea
cDepartment of Chemical Engineering, Konkuk University, 1 Hwayang-dong, Gwangjin-gu, Seoul 143-701, Republic of Korea
First published on 10th November 2014
Abstract
A simple strategy to prepare dense spherical Y2O3:Eu3+ phosphor particles with a narrow size distribution is proposed, using two steps of spray drying carried out with a commercially available spray dryer. The key idea is first to prepare hollow Y2O3:Eu3+ precursor particles by spray drying an aqueous precursor solution containing citric acid. Thereafter, nanosized particles are obtained from the precursor powders by simple ball milling and dispersed in water to form a colloidal suspension that is spray dried, forming porous granules of dense spherical nanoparticles. Next, highly crystalline Y2O3:Eu3+ is obtained by sintering the granules at temperatures above 1200 °C. The resulting Y2O3:Eu3+ particles are spherical and are shown to have good luminescence properties as a red phosphor. The feasibility of the strategy proposed here is proved experimentally.
Introduction
Phosphors play a key role in information display devices and light emitting diodes (LEDs) because their optical properties directly affect the quality of products.1–6 As display and LED technologies advance, smaller spherical phosphor particles are required with high photoluminescence characteristics.7–12 An ongoing challenge is thereby to prepare micron-sized and non-aggregated spherical phosphor powder particles with good luminescence properties.In general, commercial phosphor powders are produced by traditional solid-state reaction (SSR) methods, by mixing metal oxide precursors at high reaction temperatures for long periods of time.13–19 Although the process is relatively simple, the resulting powders have unavoidable disadvantages such as non-uniform morphologies and broad particle size distributions of about 2–20 μm. Moreover, the phosphor powders can have inhomogeneous phase distributions due to the uneven mixing of the bulky precursors during the preparation process. Flux additives are a common means to avoid these problems. Alternatively, repetitive ball milling and washing are required to prepare fine phosphor powders with good luminescence characteristics.14,18,20–22 To overcome these drawbacks, a variety of liquid- or gas-phase reaction methods have so far been proposed, including co-precipitation,23–27 sol–gel,28–34 solvothermal,35–39 combustion,40–47 spray pyrolysis,48–53 and spray drying.54–56 For industrial applications, the synthesis method should be scalable in terms of the quantity of phosphor powder required. The challenge therefore is to develop new synthetic processes using simple, commercially available production methods.
Since numerous ceramic powders have been manufactured industrially on the kilogram to ton scale by spray drying,57–64 we decided to apply this process for the large-scale synthesis of phosphor powders. However, the spray drying process has a serious drawback that the resulting powders often contain shell-like aggregates with hollow, submicron-sized primary particles.58,59,64,65 Basically, the hollow structures are the result of surface precipitation occurring first during the drying step, due to the solute concentration gradient. When phosphor particles are synthesized by a commercially available spray dryer, the formation of hollow-structured particles is inevitable. Hollowness reduces the photoluminescence and long-term stability of phosphor powders.66–69 Therefore, in view of these difficulties, it is a considerable challenging work to the development of a new strategy through which the commercial spray drying process makes it possible to the large-scale production of fine, non-agglomerated and spherical-shaped phosphor powders with narrow size distributions.
The aim of this work was to develop a simple but effectively scalable spray-drying process for the production of spherical phosphor powder particles with a narrow size distribution. To overcome the drawback of the spray drying process, we developed a new strategy, which was to use two-step spray drying. In this process, hollow and thin-shelled precursor particles are first prepared by spray drying a clear precursor solution. These hollow precursor powders are then crushed by simple ball milling, producing nano-sized particles. Next, a colloidal suspension is prepared by the crushed nanoparticles and used as the spray solution for the second spray-drying step. Finally, the resulting particles show spherical shape, dense structure, fine size, and narrow size distribution. In order to prove the viability of this strategy, europium-doped yttrium oxide (Y2O3:Eu3+) phosphor was selected as a target material because Y2O3:Eu3+ is representative of red-emitting phosphor materials in displays and fluorescent lamps.70,71 The particle formation mechanism was proposed, and the feasibility of this approach was proved with experimental evidence.
Experimental
Powder preparation
A commercial spray-drying system (Fig. S1†) was used to prepare the Y2O3:Eu3+ precursor powders with thin-walled hollow particles. The spray solution was prepared by dissolving yttrium oxide (99.9%, Rhodia) and europium oxide (99.9%, Aldrich) in distilled water containing nitric acid (60%, Aldrich). Citric acid monohydrate (CA, 99.9%, Junsei) was also added to the above solution. The total concentration of metal components in the spray solution was fixed at 0.5 M, as was the concentration of CA. The prepared spray solution was pumped into an atomizing device (20 mL min−1) in which a two-fluid nozzle was operated at a pressure of 0.8 bar in order to generate numerous droplets via a stream of hot air. The spray-dried powder was separated from the humid air by centrifugal forces in a cyclone system. The temperatures at the inlet and outlet of the spray dryer were maintained at 250 °C and 120 °C, respectively.The obtained precursor powders were calcined at 500 °C for 3 h in air, then ball milled for 5 h in deionized water to transform the hollow precursor powder into a nanoparticle suspension for a second stage of spray drying. The nanoparticle suspension was atomized as before and dried at 250 °C to produce microspheres. The powders obtained after the second spray drying were treated at temperatures between 1200 and 1400 °C for 3 h in air.
Characterization
The microstructures of the powders were observed by scanning electron microscopy (SEM, JEOL, JSM-6060) and high-resolution transmission electron microscopy (HRTEM, JEOL, JEM-2100F). The crystal phases of the powders were identified by X-ray diffraction (XRD, X'Pert PRO MPD) using Cu Kα radiation (λ = 1.5418 Å). The surface areas of the powders were obtained from N2 adsorption isotherms using Brunauer–Emmett–Teller (BET) theory. Thermogravimetric analysis was carried out in the temperature range from 25 to 650 °C using a thermogravimetric analyzer (Pyris 1 TGA, Perkin Elmer) at a heating rate of 10 °C min−1 in static air. Photoluminescence spectra were measured using a spectrophotometer (LS 50, Perkin Elmer) with a Xe flash lamp as the excitation light source. An image analyzer (ImageJ, NIH) was used to determine the particle-size distributions of the powders.Results and discussion
The key objective of this work was to obtain dense and spherical Y2O3:Eu3+ phosphor particles via a commercially available spray drying process, as shown in Scheme 1. A droplet (Scheme 1-①) is first generated through the atomization of the precursor solution through the two-phase nozzle, and the water in the droplet is dried rapidly thanks to compressed hot air (Scheme 1-②). Water evaporation results in increased salt concentration. Due to the fast drying, the salt concentration first reaches the supersaturation point at the surface of the droplets. As a result, surface precipitation begins with precursor solution still present in the inner region of droplets (Scheme 1-③). Thereby, as the evaporation proceeds, hollow particles are formed (Scheme 1-④), which are then withdrawn by a cyclone system. These hollow particles form what we call the spray-dried Y2O3:Eu3+ precursor powder. Since, citric acid (CA) was added to the spray solution as a chelating agent, the precursor particles exist as organic–inorganic composites composed of metal nitrate–citrate (Y and Eu). In order to decompose the nitrate and citrate in the precursor powders, they were calcined at 500 °C (Scheme 1-⑤). During calcination, the spray-dried organic–inorganic composites become hollow Y2O3:Eu3+ particles. Thereafter, the calcined precursor powders are disintegrated into nano-sized particles by wet ball-milling (Scheme 1-⑥), and used to prepare a colloidal suspension in water. This suspension is used as the precursor solution for the second spray drying, which produces micron-sized spherical aggregates with a porous structure consisting of dense Y2O3:Eu3+ nanoparticles (Scheme 1-⑦). Finally, following sintering, dense and highly crystalline Y2O3:Eu3+ particles are produced with a narrow size distribution (Scheme 1-⑧). This mechanism for the formation of dense spherical Y2O3:Eu3+ particles was examined experimentally as described in the following sections.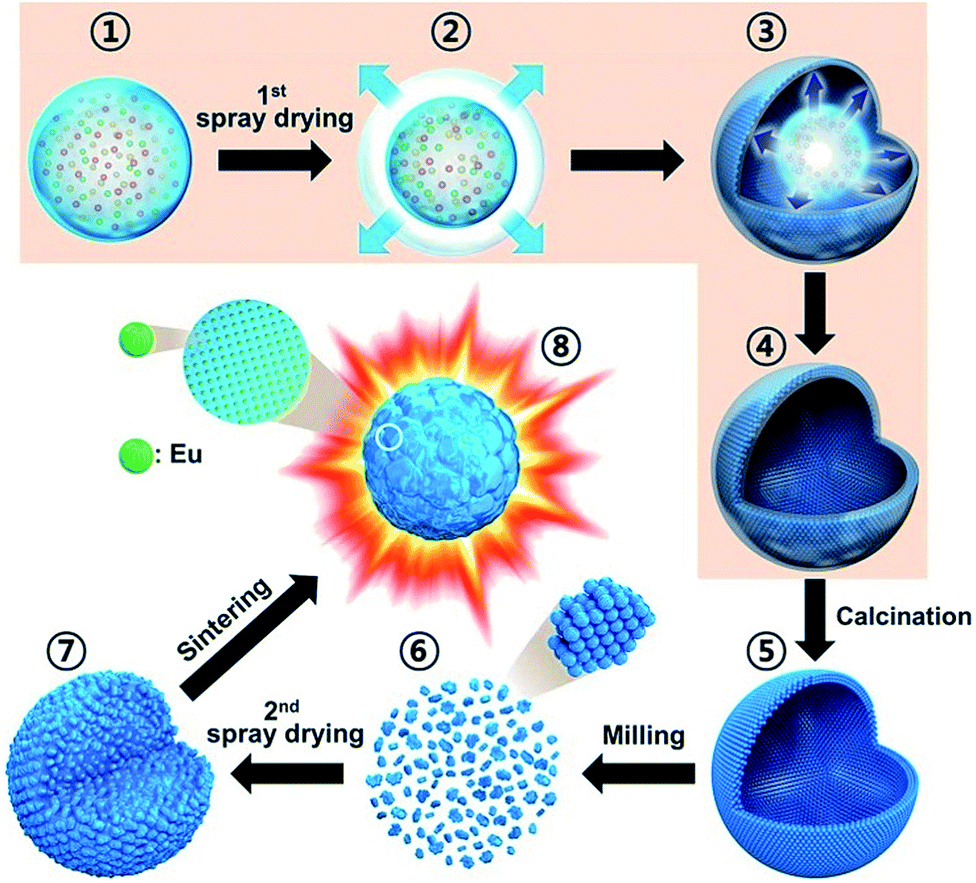 | ||
| Scheme 1 Schematic diagram of formation mechanism of the dense and spherical Y2O3:Eu3+ phosphors by using two-step spray drying process. | ||
Fig. 1(a) shows an SEM image of the Y2O3:Eu3+ precursor powders prepared by spray drying. Clearly, the as-prepared precursor particles are hollow with a thin shell. This is in good agreement with the process described in Scheme 1-④; namely, that during the process, fast drying of the droplets results in the formation of hollow particles with a thin wall composed of yttrium, europium salts and CA. Most of the spray-dried particles are larger than 10 μm, but some are fragmented. During precipitation, the first-formed shell is like the gel state of CA–metal complexes. Thereafter, the gases generated by the rapid drying of water in the interior inflate the gel droplets (Scheme 1-③). Subsequently, the evaporation of water from the gel layer generates a solid composite layer, and results in relatively large hollow particles. Highly porous shells facilitate the diffusion of gases generated on the inside, thereby maintaining the hollow structure of the particles. On the contrary, insufficient gas diffusion leads to elevated internal pressures and fragmented structures. Both these situations can occur during standard spray drying. As a result, the spray dried precursor powder in Fig. 1(a) contains both hollow spherical particles and fragmented structures.
 | ||
| Fig. 1 Morphologies and elemental mapping images of the Y2O3:Eu3+ precursor powders before and after calcination at 500 °C: (a) SEM image of spray-dried precursor powders, (b) SEM image of calcined powders, (c) TEM image of calcined powders, and (d) elemental mapping images of calcined powders. | ||
The hollow precursor particles produced in the first spray-drying step are organic–inorganic composites. To determine the optimal calcination temperature, the thermal decomposition behavior of the precursor particles was investigated by TG analysis. Fig. S2† shows the weight loss of the spray-dried Y2O3:Eu3+ precursor powder with increasing temperatures. About 11 wt% is lost up to about 100 °C due to the evaporation of adsorbed water molecules. In the temperature range from 100 to 132 °C, a weight loss of 59 wt% is observed due to the burning of CA additive in the precursor powders. Finally, a weight loss of 7 wt% occurs between 200 and 500 °C due to the decomposition of the nitrate salts and carbon residues in the spray-dried precursor powder. On the basis of the above results, 500 °C is the optimal calcination temperature to entirely eliminate the decomposable materials from the precursor powders.
The morphology and microstructure of the Y2O3:Eu3+ precursor powders were monitored by SEM (Fig. 1(b)) and TEM (Fig. 1(c)) after calcination at 500 °C. The as-prepared hollow precursor particles fragmented during calcination because their thin shell is easily broken by thermal shocks. Nevertheless, some particles having a relatively thick shell remained spherical even after calcination. As shown in Fig. 1(c), the spherical particles are hollow with a shell thickness of about 100 nm. To confirm the homogeneous distribution of Y and Eu, elemental mapping of the particles obtained after calcination was carried out, with the results shown in Fig. 1(d). These images indeed show that yttrium, oxygen and europium components are distributed uniformly over the powder particles.
After the calcination at 550 °C, the Y2O3:Eu3+ precursor powders were crushed in a planetary mill using zirconia balls to prepare nanoparticles for the second spray-drying step. Since after calcination, the micron-sized particles have a thin shell, they are readily pulverized down to nano-sized particles by simple ball milling. The TEM image of the Y2O3:Eu3+ precursor particles after ball milling in Fig. 2(a) confirms that the micron-sized precursor particles are pulverized down to a size of several tens of nanometers, which is experimental evidence for Scheme 1-⑥. Fig. 2(b) and (c) are high-resolution TEM images of the nano-sized Y2O3:Eu3+ particles. Clear lattice fringes for the (440) and (222) planes are observed, indicating that the particles are crystalline. The d spacing values of 0.27 nm and 0.31 nm correspond to the (440) and (222) planes of Y2O3 (JCPDS Card no. 43-1036). Elemental mapping images for Y, Eu, and O in the crushed nanoparticles are shown in Fig. 2(d), in indicating that all three elements are distributed uniformly in the specimen. That is, phase separation did not occur even after wet milling.
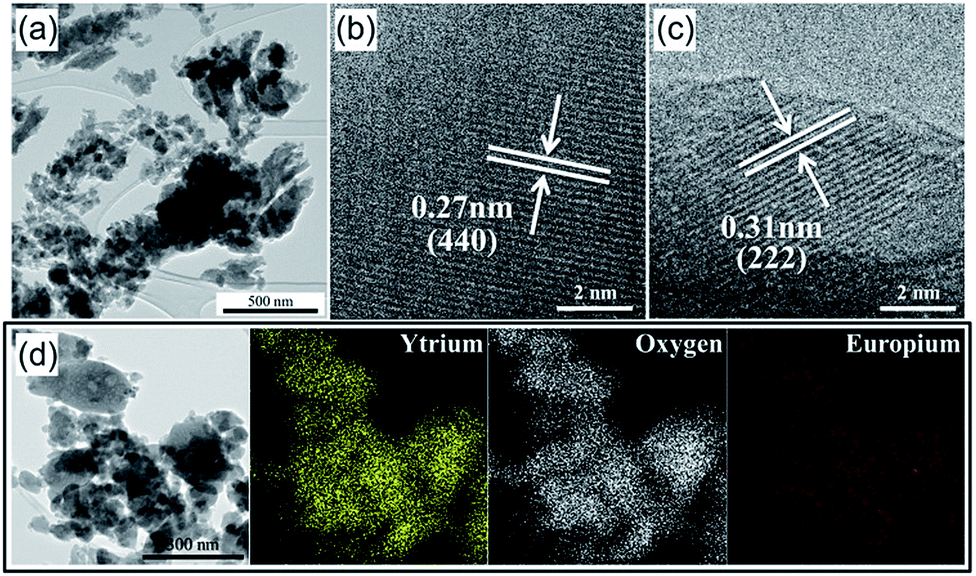 | ||
| Fig. 2 Morphologies and elemental mapping images of the crushed Y2O3:Eu3+ powders after planetary-milling process: (a) TEM image, (b) and (c) HR-TEM images, and (d) elemental mapping images. | ||
A stable colloidal suspension was prepared using the Y2O3:Eu3+ nanoparticles obtained by wet milling, which was spray dried in order to produce spherical granules several microns in size. Fig. 3 (a) shows SEM images of the prepared granules, which are aggregates of nano-sized Y2O3:Eu3+ particles. The average size of the spherical granules was 3.1 ± 0.6 μm (see Fig. S3 in the ESI†). The aggregated spherical particles obtained by spray drying the colloidal suspension do not show luminescence because Eu3+ is not entirely substituted into the Y sites of the Y2O3 host, and because of their very low crystallinity. The spray-dried granules were therefore heat treated at high temperatures to increase their crystallinity and to activate the Eu3+ ions. According to the literature on Y2O3:Eu3+ phosphor,48,50,72 a high photoluminescence was achieved for heat treatment temperatures above 1200 °C. Therefore, the granule powders obtained by two-step spray drying were heat-treated at temperatures between 1200 and 1400 °C. In Fig. 3(b–d), all the specimens have agglomeration-free spherical particles after heat treatment, regardless of the sintering temperature. With increasing sintering temperatures however, the particles become gradually smaller and denser, and Fig. S2† shows that the particle size distributions become narrower. To investigate variations in porosity, specific surface areas were measured using nitrogen adsorption isotherms (displayed in Fig. S4†) according to BET theory. The resulting BET surface areas were 5.3, 3.1, and 1.5 m2 g−1 for sintering temperatures of 1200, 1300, and 1400 °C, respectively. The reduction in specific surface area from 5.3 m2 g−1 to 1.5 m2 g−1 for sintering temperatures from 1200 °C to 1400 °C, indicates that densification occurred. This densification can be connected to a reduction in average particle size with increased sintering temperatures. For sintering temperatures of 1200, 1300 and 1400 °C, the average particle size was 2.5 ± 0.5, 2.1 ± 0.5, and 1.7 ± 0.3 μm, respectively.
 | ||
| Fig. 3 SEM images of the (a) Y2O3:Eu3+ powders prepared by second-step spray drying process, and post-treated Y2O3:Eu3+ powders at the temperature of (b) 1200 °C, (c) 1300 °C, and (d) 1400 °C. | ||
X-ray diffraction patterns were measured in order to record the phases present in the samples as a function of the sintering temperatures. Fig. S5† indicates the presence of pure cubic phase irrespective of the sintering temperature, and no transition to the monoclinic phase was observed. The mean crystallite sizes, calculated from the peak widths of the (222) reflection in the XRD patterns using Scherrer's equation, were 48, 108, and 161 nm for sintering temperatures of 1200, 1300, and 1400 °C, respectively. The crystallite size increased with increasing sintering temperatures.
Fig. 4 shows the photoluminescence properties of Y2O3:Eu3+ phosphor powders obtained after sintering at various temperatures. The emission spectra were measured under 254 nm ultraviolet (UV) light, while the excitation spectra were measured at the main emission peak of 611 nm. It is well known that cubic Y2O3:Eu3+ phosphor absorbs UV light through a charge transfer band (CTB) and emits red light at 611 nm due to the 5D0–7F2 transition of Eu3+ ions.73 The excitation band at around 240–260 nm is attributed to CTB absorption which consists of two peaks as shown in the inset of Fig. 4(a). The CTB peak position depends on the particle size.74 A blue shift in CTB positions occurs because the ionicity between oxygen and Eu3+ decreases due to the decrease in the covalency as reducing the particle size below several tens of nanometers. As shown in Fig. 3, the micron-sized Y2O3:Eu3+ particles are agglomerates consisting of nano-sized primary particles. Thus, the synthesized Y2O3:Eu3+ particles show two split excitation spectra peaking at about 248 nm and 262 nm for primary nanoparticles and micron-sized bulk particles, respectively. As increasing the calcination temperature, the primary particle sizes become larger, which is responsible for the increase in the peak intensity at 262 nm. The red emission observed is well matched with the luminescence characteristics of Y2O3:Eu3+ phosphor. Under the 254 nm excitation, the highest photoluminescence intensity for the post-treated Y2O3:Eu3+ phosphor powders was achieved at 1200 °C, in agreement with the literature.50,72 The photoluminescence intensity of the Y2O3:Eu3+ powder post-treated at 1200 °C was 105% of that of the powder post-treated at 1400 °C. The excitation and emission spectra of the commercial Y2O3:Eu3+ phosphor powders were compared with those of the synthesized phosphor powders in Fig. S6.† Thus, no difference in the emission peak position was observed for all prepared samples as well as the commercial one. However, the synthesized Y2O3:Eu3+ phosphor powders had lower emission intensity (about 62%) than that of the commercial product. The main reason of the low emission intensity of the Y2O3:Eu phosphor powders prepared by two-step spray drying method was contamination of the powders during the milling process of the precursor powders for two-step spray drying. In addition, the low emission intensity can be improved by optimizing Eu3+ concentration and using flux materials usually used in the production of commercial phosphor via a solid-state method. These results prove that this new approach based on commercially available spray drying equipment is successful in producing Y2O3:Eu3+ red phosphor with good characteristics, notably the small size, spherical shape, dense structure and narrow size distribution of the powder particles. Furthermore, this method is easily scalable and can therefore be applied to the mass production of phosphor particles.
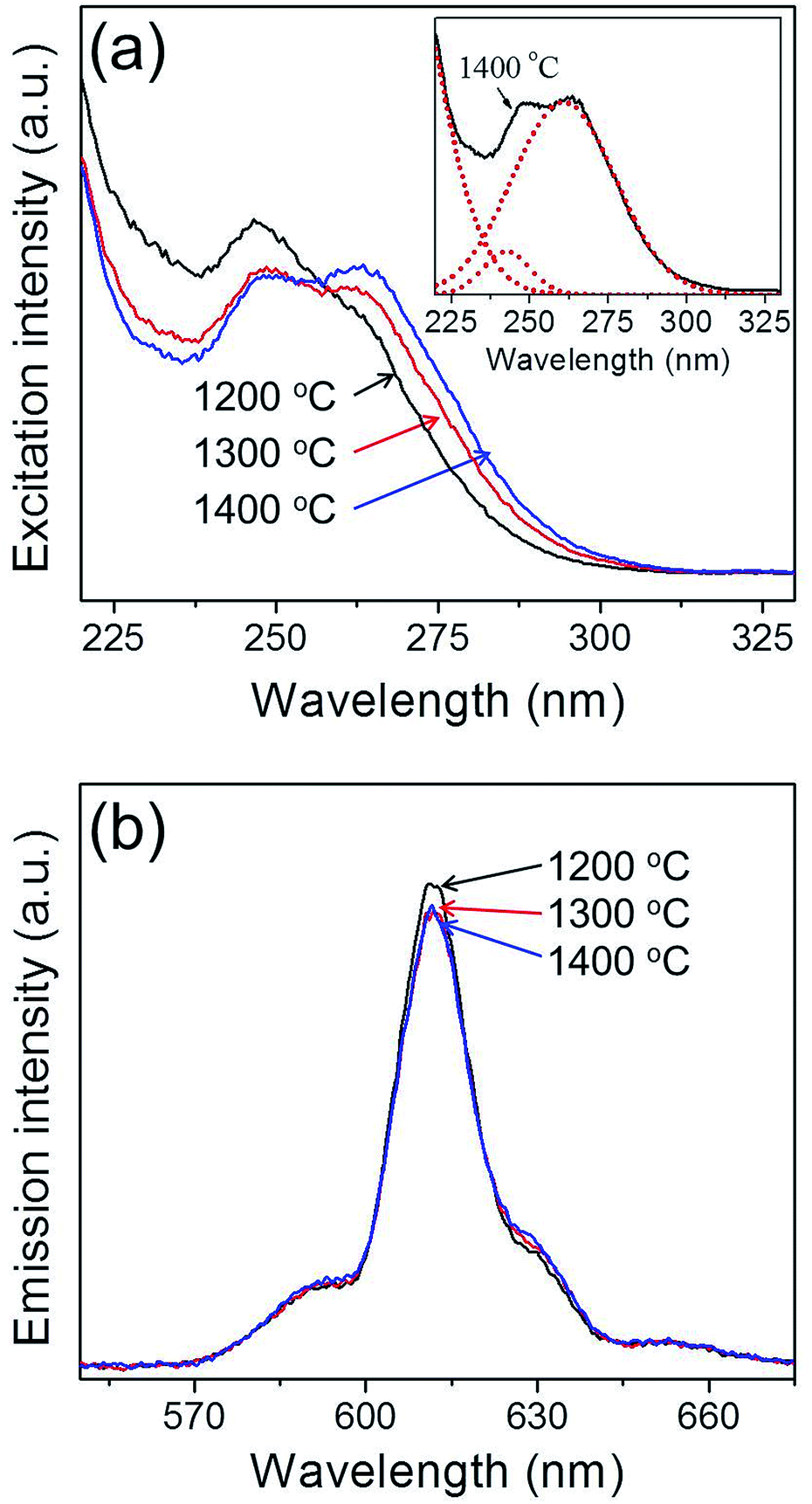 | ||
| Fig. 4 Photoluminescence (a) excitation, and (b) emission spectra of the Y2O3:Eu3+ phosphor powders formed at the various sintering temperatures. | ||
Conclusions
Dense micron-sized phosphor particles with a narrow size distribution were successfully prepared by using a commercially available spray drying system. A new strategy to obtain dense spherical particles via spray drying was suggested and its feasibility was proved experimentally. Hollow Y2O3:Eu3+ precursor particles were obtained by spray drying a clear solution containing nitrate precursor and citric acid used as an organic additive. The prepared precursor powder thereby contained organic–inorganic composites. According to the thermo gravimetric analysis, all the organic components were removed by calcination at 500 °C. Following calcination, the hollow precursor particles were readily pulverized down to the nano-size via simple wet ball-milling. Transmission electron microscopy indicated that these particles were several tens of nanometers in size, and according to elemental mapping, were uniformly covered with Y, Eu, and O, without any phase separation. Micron-sized granules consisting of nanoparticles were obtained by spray drying a colloidal suspension prepared by dispersing the nano-sized precursor particles in water. Finally, the nanoparticle aggregates were sintered at high temperature in order to achieve a high degree of crystallinity and to activate the Eu3+ ions. The resulting Y2O3:Eu3+ particles were dense and spherical, with an average size of 1.7–2.5 μm and good photoluminescence characteristics as a red phosphor.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (no. 2012R1A2A2A02046367).Notes and references
- R. Zhang, H. Lin, Y. Yu, D. Chen, J. Xu and Y. Wang, Laser Photonics Rev., 2014, 8, 158 CrossRef CAS.
- P. Pust, V. Weiler, C. Hecht, A. Tücks, A. S. Wochnik, A.-K. Henß, D. Wiechert, C. Scheu, P. J. Schmidt and W. Schnick, Nat. Mater., 2014, 13, 891 CrossRef CAS PubMed.
- N. N. Dong, M. Pedroni, F. Piccinelli, G. Conti, A. Sbarbati, J. E. Ramírez-Hernández, L. M. Maestro, M. C. Iglesias-de la Cruz, F. Sanz-Rodriguez and A. Juarranz, ACS Nano, 2011, 5, 8665 CrossRef CAS PubMed.
- C. Ronda, Prog. Electromagn. Res., 2014, 147, 81 Search PubMed.
- K. N. Shinde and S. J. Dhoble, Crit. Rev. Solid State Mater. Sci., 2014, 39, 459 CrossRef CAS.
- Q. Tan, J. Li and X. Zeng, Crit. Rev. Environ. Sci. Technol., 2014 DOI:10.1080/10643389.2014.900240.
- T. Igarashi, M. Ihara, T. Kusunoki, K. Ohno, T. Isobe and M. Senna, Appl. Phys. Lett., 2000, 76, 1549 CrossRef CAS PubMed.
- Y. C. Kang and H. D. Park, Appl. Phys. A, 2003, 77, 529 CrossRef CAS PubMed.
- C. Panatarani, I. W. Lenggoro and K. Okuyama, J. Phys. Chem. Solids, 2004, 65, 1843 CrossRef CAS PubMed.
- Y. C. Kang, I. W. Lenggoro, S. B. Park and K. Okuyama, J. Solid State Chem., 1999, 146, 168 CrossRef CAS.
- Y. C. Kang, H. S. Roh, S. B. Park and K. Y. Jung, Jpn. J. Appl. Phys., 2004, 43, 5302 CrossRef CAS.
- N. Joffin, B. Caillier, J. Dexpert-Ghys, M. Verelst, G. Baret, A. Garcia, P. Guillot, J. Galy, R. Mauricot and S. Schamm, J. Phys. D: Appl. Phys., 2005, 38, 3261 CrossRef CAS.
- C. Zhao, X. Yin, Y. Wang, F. Huang and Y. Hang, J. Lumin., 2012, 132, 617 CrossRef CAS PubMed.
- H. K. Yang and J. H. Jeong, J. Phys. Chem. C, 2009, 114, 226 Search PubMed.
- H. Y. Chen, R. Y. Yang and S. J. Chang, J. Phys. Chem. Solids, 2013, 74, 344 CrossRef CAS PubMed.
- Y. Qiao, X. Zhang, X. Ye, Y. Chen and H. Guo, J. Rare Earths, 2009, 27, 323 CrossRef.
- Z. Xiaoxia, W. Xiaojun, C. Baojiu, M. Qingyu, D. Weihua, R. Guozhong and Y. Yanmin, J. Rare Earths, 2007, 25, 15 CrossRef.
- H. Guo, X. Wang, X. Zhang, Y. Tang, L. Chen and C. Ma, J. Electrochem. Soc., 2010, 157, J310 CrossRef CAS PubMed.
- J. H. Lee and Y. J. Kim, Mater. Sci. Eng., B, 2008, 146, 99 CrossRef CAS PubMed.
- L. Zhang, Z. Lu, P. Han, J. Lu, N. Xu, L. Wang and Q. Zhang, J. Am. Ceram. Soc., 2012, 95, 3871 CrossRef CAS PubMed.
- Y. M. Peng, Y. K. Su and R. Y. Yang, Mater. Res. Bull., 2013, 48, 1946 CrossRef CAS PubMed.
- T. Y. Choi, Y. H. Song, H. R. Lee, K. Senthil, T. Masaki and D. H. Yoon, Mater. Sci. Eng., B, 2012, 177, 500 CrossRef CAS PubMed.
- J. G. Fan, S. Q. Sun and X. H. Ma, Chem. Ind. Eng., 2007, 24, 497 CAS.
- K. Zhang, H. Z. Liu, Y. T. Wu and W. B. Hu, J. Alloys Compd., 2008, 453, 265 CrossRef CAS PubMed.
- W. Pan, G. Ning, X. Zhang, J. Wang, Y. Lin and J. Ye, J. Lumin., 2008, 128, 1975 CrossRef CAS PubMed.
- J. Chen, Y. Shi and J. Shi, J. Mater. Res., 2004, 19, 3586 CrossRef CAS.
- J. G. Li, T. Ikegami, J. H. Lee, T. Mori and Y. Yajima, J. Eur. Ceram. Soc., 2000, 20, 2395 CrossRef CAS.
- V. R. Bandi, B. K. Grandhe, K. Jang, H. S. Lee, D. S. Shin, S. S. Yi and J. H. Jeong, J. Alloys Compd., 2012, 512, 264 CrossRef CAS PubMed.
- Y. Xie, L. J. Xiao, F. Q. Yan, Y. J. Chen, W. Z. Li and X. J. Geng, J. Nanosci. Nanotechnol., 2014, 14, 4486 CrossRef CAS PubMed.
- M. K. Chong, K. Pita and C. H. Kam, J. Phys. Chem. Solids, 2005, 66, 213 CrossRef CAS PubMed.
- L. Xiao, Q. Xiao and Y. Liu, J. Rare Earths, 2011, 29, 39 CrossRef CAS.
- H. Wu, Y. Hu, W. Zhang, F. Kang, N. Li and G. Ju, J. Sol–Gel Sci. Technol., 2012, 62, 227 CrossRef CAS PubMed.
- Q. Chen, Y. Shi, S. Wang, J. Chen and J. Shi, J. Eur. Ceram. Soc., 2007, 27, 191 CrossRef CAS PubMed.
- J. Liao, H. You, D. Zhou, H.-r. Wen and R. Hong, Opt. Mater., 2012, 34, 1468 CrossRef CAS PubMed.
- Y. Song, H. You, Y. Huang, M. Yang, Y. Zheng, L. Zhang and N. Guo, Inorg. Chem., 2010, 49, 11499 CrossRef CAS PubMed.
- C. T. Lee, F. S. Chen and C. H. Lu, J. Alloys Compd., 2010, 490, 407 CrossRef CAS PubMed.
- Z. Zhu, D. Liu, H. Liu, G. Li, J. Du and Z. He, J. Lumin., 2012, 132, 261 CrossRef CAS PubMed.
- S. Kumar, Z. Jindal, N. Kumari and N. K. Verma, J. Nanopart. Res., 2011, 13, 5465 CrossRef CAS.
- M. K. Devaraju, S. Yin and T. Sato, J. Nanosci. Nanotechnol., 2010, 10, 731 CrossRef CAS PubMed.
- J. T. Ingle, R. P. Sonekar, S. K. Omanwar, Y. Wang and L. Zhao, Combust. Sci. Technol., 2014, 186, 83 CrossRef CAS.
- S. Ekambaram and M. Maaza, J. Alloys Compd., 2005, 395, 132 CrossRef CAS PubMed.
- I. M. Nagpure, K. N. Shinde, V. Kumar, O. M. Ntwaeaborwa, S. J. Dhoble and H. C. Swart, J. Alloys Compd., 2010, 492, 384 CrossRef CAS PubMed.
- X. Qian, X. Pu, D. Zhang, L. Li, M. Li and S. Wu, J. Lumin., 2011, 131, 1692 CrossRef CAS PubMed.
- Y. P. Fu, J. Mater. Sci., 2007, 42, 5165 CrossRef CAS PubMed.
- X. Luo, W. Cao and M. Xing, J. Rare Earths, 2006, 24, 20 CrossRef.
- Y. Zhou, Y. Yoshizawa, K. Hirao, Z. Lenčéš and P. Šajgalík, J. Eur. Ceram. Soc., 2011, 31, 151 CrossRef CAS PubMed.
- F. Hong, L. Zhou, L. Li, Q. Xia and X. Luo, Opt. Commun., 2014, 316, 206 CrossRef CAS PubMed.
- Y. C. Kang, H. S. Roh and S. B. Park, Adv. Mater., 2000, 12, 451 CrossRef CAS.
- K. Y. Jung and Y. C. Kang, Mater. Lett., 2004, 58, 2161 CrossRef CAS PubMed.
- J. S. Cho, K. M. Yang and Y. C. Kang, CrystEngComm, 2014, 16, 6170 RSC.
- Y. C. Kang, S. B. Park, I. W. Lenggoro and K. Okuyama, J. Electrochem. Soc., 1999, 146, 2744 CrossRef CAS PubMed.
- L. Mancic, K. Marinkovic, B. A. Marinkovic, M. Dramicanin and O. Milosevic, J. Eur. Ceram. Soc., 2010, 30, 577 CrossRef CAS PubMed.
- L. S. Wang, Y. H. Zhou, Z. W. Quan and J. Lin, Mater. Lett., 2005, 59, 1130 CrossRef CAS PubMed.
- W. D. Wu, R. Amelia, N. Hao, C. Selomulya, D. Zhao, Y. L. Chiu and X. D. Chen, AIChE J., 2011, 57, 2726 CrossRef CAS.
- T. Han, S. Cao, D. Zhu, C. Zhao, M. Ma, M. Tu and J. Zhang, Optik, 2013, 124, 3539 CrossRef CAS PubMed.
- A. Nakamura, N. Nambu and H. Saitoh, Sci. Technol. Adv. Mater., 2005, 6, 210 CrossRef CAS PubMed.
- G. Jean, V. Sciamanna, M. Demuynck, F. Cambier and M. Gonon, Ceram. Int., 2014, 40, 10197 CrossRef CAS PubMed.
- G. Bertrand, P. Roy, C. Filiatre and C. Coddet, Chem. Eng. Sci., 2005, 60, 95 CrossRef CAS PubMed.
- S. J. Lukasiewicz, J. Am. Ceram. Soc., 1989, 72, 617 CrossRef CAS PubMed.
- K. Uematsu, J. Y. Kim, M. Miyashita, N. Uchida and K. Saito, J. Am. Ceram. Soc., 1990, 73, 2555 CrossRef CAS PubMed.
- M. Vicent, E. Sánchez, I. Santacruz and R. Moreno, J. Eur. Ceram. Soc., 2011, 31, 1413 CrossRef CAS PubMed.
- D. S. Jung, T. H. Hwang, S. B. Park and J. W. Choi, Nano Lett., 2013, 13, 2092 CrossRef CAS PubMed.
- M. Serantoni, A. Piancastelli, A. L. Costa and L. Esposito, Opt. Mater., 2012, 34, 995 CrossRef CAS PubMed.
- H. Bian, Y. Yang, Y. Wang and W. Tian, Powder Technol., 2012, 219, 257 CrossRef CAS PubMed.
- M. R. Loghman-Estarki, M. Pourbafrany, R. Shoja Razavi, H. Edris, S. R. Bakhshi, M. Erfanmanesh, H. Jamali, S. N. Hosseini and M. Hajizadeh-Oghaz, Ceram. Int., 2014, 40, 3721 CrossRef CAS PubMed.
- Y. C. Kang, H. S. Roh and S. B. Park, J. Am. Ceram. Soc., 2001, 84, 447 CrossRef CAS PubMed.
- E. J. Kim, Y. C. Kang, H. D. Park and S. K. Ryu, Mater. Res. Bull., 2003, 38, 515 CrossRef CAS.
- X. Li, Q. Li, J. Wang and S. Yang, Mater. Sci. Eng., B, 2006, 131, 32 CrossRef CAS PubMed.
- J. Zhou, Y. Wang, B. Liu and J. Liu, J. Phys. Chem. Solids, 2011, 72, 995 CrossRef CAS PubMed.
- M. K. Devaraju, S. Yin and T. Sato, J. Cryst. Growth, 2009, 311, 580 CrossRef CAS PubMed.
- X. Hou, S. Zhou, Y. Li and W. Li, J. Alloys Compd., 2010, 494, 382 CrossRef CAS PubMed.
- K. Y. Jung, C. H. Lee and Y. C. Kang, Mater. Lett., 2005, 59, 2451 CrossRef CAS PubMed.
- G. Blasse and B. C. Geiabmaier, Luminescent Materials, Springer, Berlin, 1994 Search PubMed.
- T. Igarashi, M. Ihara, T. Kusunoki, K. Ohno, T. Isobe and M. Senna, Appl. Phys. Lett., 2000, 76, 1549 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09225b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |