Direct conversion of cellulose using carbon monoxide and water on a Pt–Mo2C/C catalyst†
Jing
Li
a,
Lingtao
Liu
a,
Yue
Liu
a,
Mingzhe
Li
a,
Yihan
Zhu
b,
Haichao
Liu
a,
Yuan
Kou
a,
Jizhe
Zhang
b,
Yu
Han
*b and
Ding
Ma
*a
aBeijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, Beijing, China. E-mail: dma@pku.edu.cn; Fax: +86 10 6275 8603; Tel: +86 10 6275 8603
bAdvanced Membranes and Porous Materials Center, Physical Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal, Saudi Arabia. E-mail: yu.han@kaust.edu.sa; Tel: +966 2 808 2407
First published on 21st October 2013
Abstract
CO and H2O were employed as the hydrogen source for cellulose conversion to polyols. Pt–Mo2C/C tandem catalyst with the Pt–Mo2C domain responsible for H2 and/or H production and the Pt–C domain for cellulose conversion was fabricated. Considerable polyols were obtained over this tandem Pt–Mo2C/C catalyst.
Broader contextUnlike the traditional cellulose conversion to polyols processes which employed high pressure H2, we utilized the hydrogen in water via a CO–H2O–cellulose reaction system. In this hydrogen-free reaction system, good catalytic performance with relatively high yield towards polyols was obtained over a Pt–Mo2C/C catalyst. By using HAADF-STEM, XPS and TPD methods, it was concluded that the most active Pt–Mo2C/C catalyst was a bifunctional nanostructure, with the Pt–Mo2C domain responsible for the hydrogen species formation while the Pt–C domain was responsible for the consequent cellulose transformation. Although the reaction parameters are still subject to optimization, our findings provide not only a new base for the design of next generation cellulose activation catalysts, but also a new strategy for the development of green and sustainable chemistry in general. |
Cellulosic biomass is a sustainable and climate-friendly alternative to fossil-based raw materials.1 However, very few processes for converting cellulosic biomass directly into value-added products have been practically successful because such processes rely on effectively breaking strong hydrogen-bond networks as well as selectively cleaving designated C–C and/or C–O bonds.2 A few small-scale approaches were reported for catalytic conversion of cellulose to hydrocarbons,3 bio-oils,4 hydrogen,5 syngas,6 and fine chemicals,7 but novel, green and cost-effective processes that are conducted under relatively mild reaction conditions remain to be developed. Since the landmark works by Fukuoka et al.8 and Yan et al.9 showing that cellulose can be efficiently converted to polyols by noble metal catalysts, many excellent catalytic strategies have been developed for selective cellulose conversion via catalytic hydrogenation10 and especially by elegant methods for the treatment of the native cellulose.10d,e,g Of particular interest is the elegant route from cellulose to ethylene glycol achieved by Zhang et al.10b in which tungsten is a key to the excellent selectivity. Significantly, besides a fraction of the reactions that were conducted in ionic liquid or other organic phases,11 most of the cellulose conversion processes developed so far were realized in the aqueous phase. In these processes, water serves as a green solvent and also participates in the hydrolysis of 1,4-β-glycosidic bonds, leading to the formation of glucose, which is an important intermediate for further hydrogenation-assisted transformation reactions. Although the hydrogenation of cellulose initiated a new approach to biomass valorization, high hydrogen (H2) pressures are needed due to the low solubility of hydrogen in water, and H2 insufficiency may cause undesired side reactions such as cracking and coking.
Given that the water-gas shift (CO + H2O → CO2 + H2, WGS) reaction is a well-established industrial process for large-scale hydrogen production, we reasoned that carbon monoxide (CO) and liquid water may be employed as an expedient alternative source of hydrogen gas for catalytic conversion of cellulose. The use of CO–H2O-mediated reduction has advantages over conventional hydrogenation reactions, obviating the need for external hydrogen produced from either a feedstock or power derived from fossil sources. In addition to the obvious economic benefit, a hydrogen-free CO–H2O-mediated reductive transformation can even run under mild pressure in an inert gas atmosphere, leading to improved operational safety.12 However, the implementation of a truly convenient and cost-competitive protocol for cellulose conversion remains challenging, partly due to the slow reaction rate of the WGS reaction, although it is thermodynamically favored at low temperature (K1073K = 1.105, K300K = 1.5 × l03). Here, we describe an effective Pt–Mo2C/C catalyst for cellulose conversion without the use of molecular hydrogen. We conduct the reaction at 250 °C with CO and H2O as the hydrogen source to convert the cellulose extremely quickly to various desired products. Our investigation reveals that the Pt–Mo2C/C catalyst is actually a tandem catalytic system, with the Pt–Mo2C domains responsible for the generation of surface/gas-phase hydrogen-species and the Pt–C domains responsible for the subsequent cellulose hydrogenation/hydrogenolysis reactions.
We prepared a series of Pt and Mo2C-based catalysts including Pt/C, Mo2C/C, Pt/Mo2C, and Pt–Mo2C/C using a temperature-programmed reduction (TPR) method (see ESI†). The three Mo2C-containing catalysts were passivated in 0.5% O2–N2 overnight at room temperature before characterization and catalytic testing. For simplicity, we call the passivated catalysts Mo2C/C, Pt–Mo2C/C and Pt/Mo2C unless otherwise stated. The X-ray diffraction (XRD) profiles in Fig. 1a show that Mo2C/C, Pt–Mo2C/C and Pt/Mo2C contained hexagonal compact phase (hcp) β-Mo2C. This result indicated that the TPR method was effective in the fabrication of a carbide catalyst. While diffraction reflections of metallic Pt (2θ = 39.7° (111), 46.2° (200) and 67.4° (220)) were observed for the Pt/C catalyst, they were very weak for both the Pt/Mo2C and Pt–Mo2C/C catalysts, because the Pt particles were well dispersed with a very small crystal size (as confirmed by STEM, Fig. 2). At the same time, XRD confirmed that no bulk molybdenum oxides were formed in the samples. X-ray photoelectron spectroscopy (XPS) experiments provided information about the surface states of the catalysts. In the C1s XPS region (Fig. 1b), a carbidic carbon peak at around 282.8 eV (ref. 13) was detected for the Mo2C/C, Pt–Mo2C/C and Pt/Mo2C catalysts, confirming the formation of molybdenum carbide in these catalysts. Accordingly, carbidic Mo 3d5/2 peaks at 227.7 eV (ref. 14) were observed for all the Mo2C-containing samples, although they were in the company of shoulder peaks from molybdenum oxide at a higher binding energy (Fig. 1c).15 This suggests that the passivation process resulted in the partial surface oxidation of molybdenum carbide.15,16 The Pt/C catalyst had a Pt 4f7/2 peak at 71.0 eV (ESI, Fig. S1†), which is assigned to metallic Pt.17 For Pt on Pt/Mo2C, the Pt 4f7/2 peak was 70.7 eV. A 0.5 eV high energy shift was observed on the Pt–Mo2C/C. The reasons for this energy shift are not well understood yet, but these different electronic structures of Pt should impact the reaction activities.
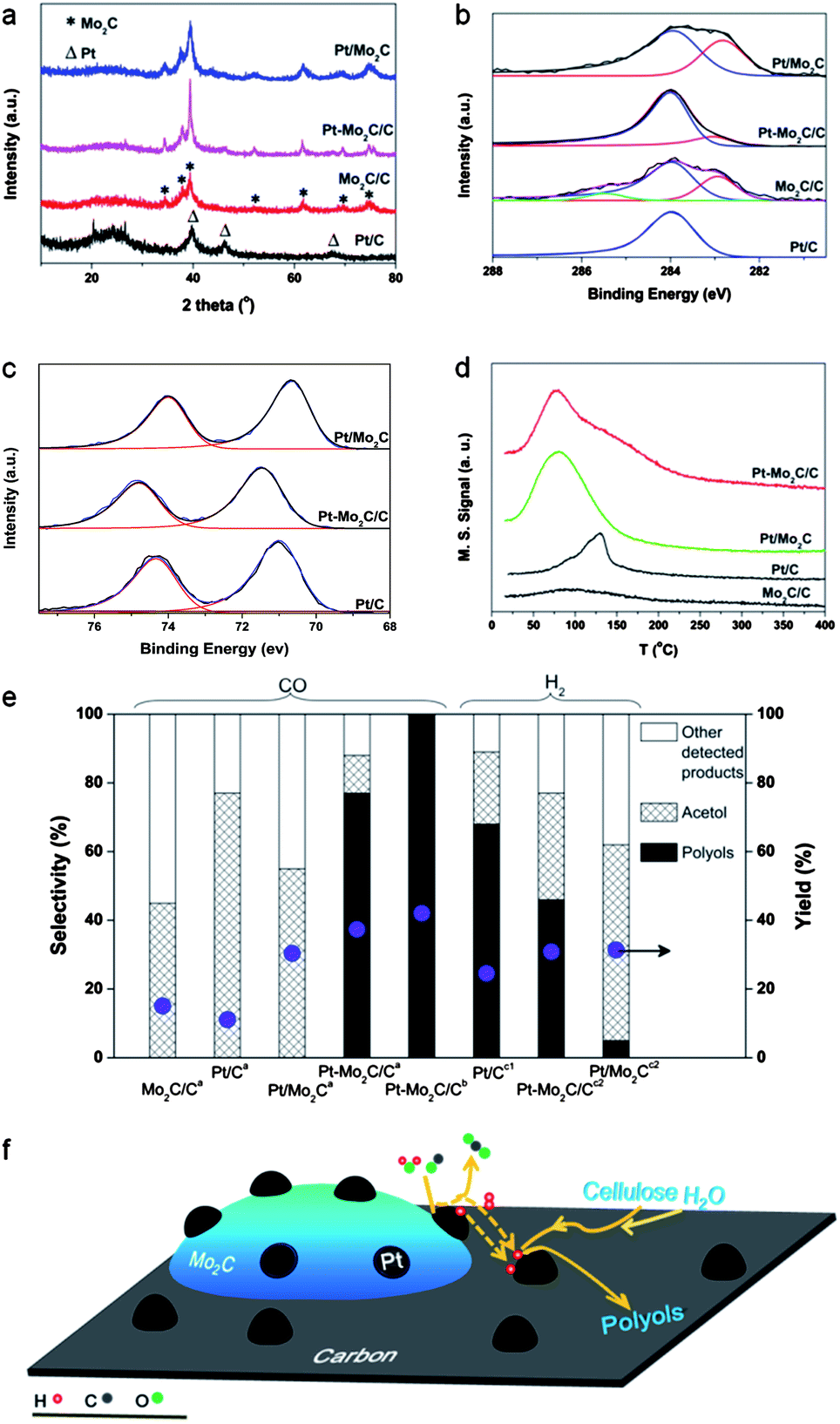 | ||
| Fig. 1 (a) XRD patterns of the passivated samples. (b) C1s XPS spectra of the passivated samples. (c) Mo3d XPS spectra of the passivated samples. (d) TPD profiles of CO desorption (m/z = 28 signals) on the samples. (e) Detected products distribution. Yields and selectivities were calculated based on carbon. The blue dots represent the total yield of the products. Other products include 2,5-hexanedione, 2-methyl-2-cyclopenten-1-one, 3-methyl-2-cyclopenten-1-one, furfural, and HMF. aPassivated catalysts, 4.5 MPa CO, 250 °C, 15 min. bUnpassivated catalyst, 4.5 MPa CO, 240 °C, 30 min. c1Passivated catalyst, 3.7 MPa H2, 250 °C, 15 min.c2Passivated catalysts, 4.5 MPa H2, 250 °C, 15 min. (f) Schematic illustration of cellulose conversion over the Pt–Mo2C/C tandem catalyst with CO and H2O as the hydrogen source. | ||
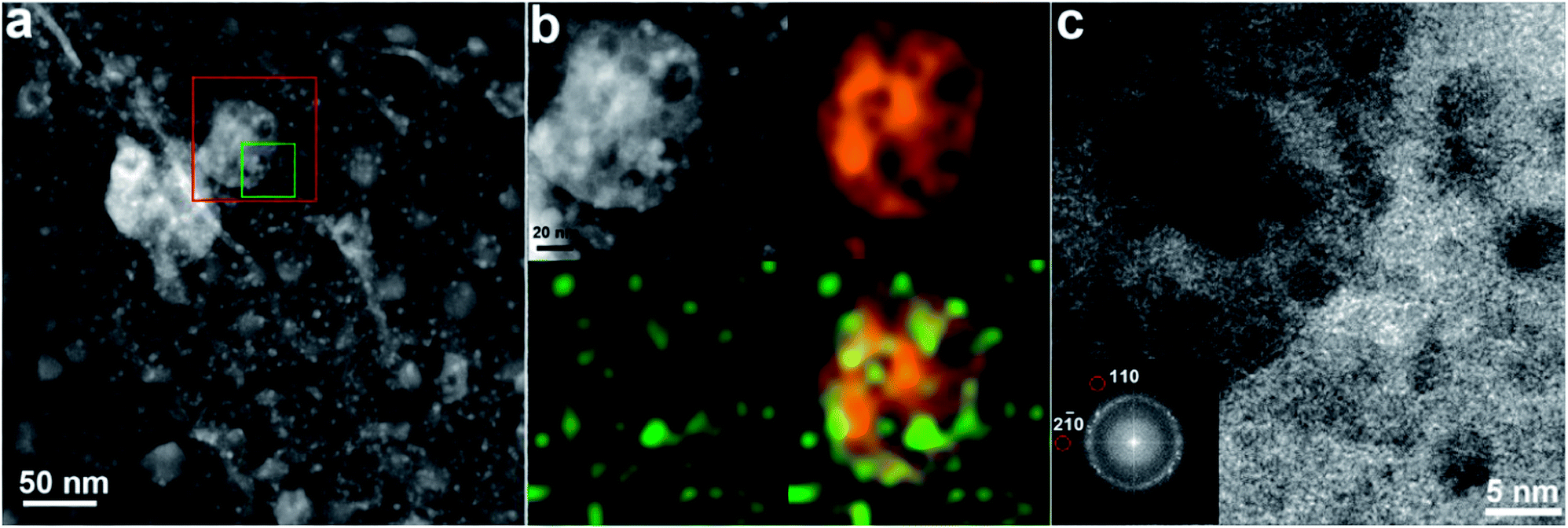 | ||
| Fig. 2 (a) A high-angle annular dark-field (HAADF)-STEM image of the Pt–Mo2C/C composite catalyst, showing two types of particles of different sizes; the large particles with a broad size distribution (20–50 nm) and the small particles with relatively uniform sizes (∼5 nm) correspond to Mo2C and Pt, respectively. (b) A magnified HAADF-STEM image (upper-left) of the red square region in (a) and the corresponding EDX elemental mappings of Mo (upper-right) and Pt (lower-left). The combined mapping image is shown in the lower right corner. (c) A HRTEM image of the region in the green square in (a); the corresponding Fast Fourier Transform (FFT) diffractogram (inset) confirms the co-existence of Mo2C and Pt crystals, in which the red circles highlight the reflections associated with Mo2C. | ||
Cellulose conversion reactions were conducted in the aqueous phase at 250 °C under 4.5 MPa CO. As shown in Table 1, Pt/C, Mo2C/C and Pt/Mo2C catalysts were not able to convert cellulose to polyols, but certain amounts of acetol and other minor products were detected. It is well known that in water-based cellulose transformation processes, hot water under high pressure will generate protons to facilitate the hydrolysis of 1,4-glycosidic bonds of cellulose with the help of metal/alumina or metal/carbon catalysts. This is one of the key steps in the depolymerization of cellulose into glucose. The obtained glucose then undergoes isomerization into fructose and subsequently retro-aldol condensation followed by hydrogenation, leading to the formation of acetol (Scheme S1†). We found that more acetol (yield: 16.7%) was produced on the Pt/Mo2C catalyst than on the Mo2C/C (6.8%) and Pt/C (8.5%) catalysts (Table 1). As indicated by XPS, the surface of Mo2C can be partially oxidized during the passivation process to molybdenum oxycarbide to provide additional Lewis acid sites.18 Because Lewis acid sites accelerate the isomerization of glucose to fructose19 as well as the retro-aldol condensation process,20 it is reasonable that the Pt/Mo2C composite catalyst with the highest Mo2C population could yield more acetol than did its mono-metal counterparts.
| Entry | Catalyst | Reaction atmosphere | WGS rateb/molCO molPt−1 s−1 | Yieldc/% | Polyols distributionc/% | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Polyols | Acetol | 2,5-HD/3-MCPh | 2-MCPh | Furfural/HMF | EGh | 1,2-PDh | 1,2-BDh | 1,2-HDh | Glycerol | Erythritol | Sorbitol | Mannitol | ||||
| a Reaction conditions: 60 ml water, 0.5 g cellulose, 0.15 g catalyst. Metal loadings are determined by inductively coupled plasma-atomic emission spectrometry. b 0.15 g passivated catalyst, 4.5 MPa CO, 250 °C, 5 min. c Yields and selectivities were calculated based on carbon. d Passivated catalysts, 4.5 MPa, 250 °C, 15 min. e The catalysts were used without passivation, 4.5 MPa CO, 240 °C, 30 min. f Passivated catalysts, 3.7 MPa H2, 250 °C, 15 min. g Passivated catalysts, 4.5 MPa H2, 250 °C, 15 min. h Abbreviations: EG for ethylene glycol, 1,2-PD for 1,2-propanediol, 1,2-BD for 1,2-butanediol, 1,2-HD for 1,2-hexanediol, 2,5-HD for 2,5-hexanedione, 2-MCP for 2-methyl-2-cyclopenten-1-one, and 3-MCP for 3-methyl-2-cyclopenten-1-one. | ||||||||||||||||
| 1 | 48%Mo2C/Cd | CO | 0 | 0 | 6.8 | 1.1 | 2.3 | 4.9 | — | — | — | — | — | — | — | — |
| 2 | 7%Pt/Cd | CO | 0.04 | 0 | 8.5 | <0 | 2.6 | <1 | — | — | — | — | — | — | — | — |
| 3 | 7%Pt/Mo2Cd | CO | 2.19 | <1 | 16.7 | 8.2 | 5.5 | <1 | — | — | — | — | — | — | — | — |
| 4 | 7%Pt–40%Mo2C/Cd | CO | 3.54 | 28.7 | 4.0 | 3.1 | 1.6 | 0 | 39.4 | 32.0 | 15.0 | 13.6 | — | — | — | — |
| 5 | 7%Pt–40%Mo2C/Ce | CO | 42.1 | 0 | <1 | 0 | 0 | 5.9 | 11.2 | 3.3 | 12.1 | <1 | 7.6 | 51.6 | 8.3 | |
| 6 | 7%Pt–40%Mo2C/Cd | N2 | 0 | 8.9 | 4.6 | 4.0 | 0 | — | — | — | — | — | — | — | — | |
| 7 | 7%Pt/Cf | H2 | 16.7 | 5.2 | 2.7 | <1 | 0 | 24.6 | 18.0 | 8.4 | 24.6 | — | — | 24.6 | <1 | |
| 8 | 7%Pt/Mo2Cg | H2 | 1.5 | 18.0 | 7.5 | 4.4 | <1 | 100 | — | — | — | — | — | — | — | |
| 9 | 7%Pt–40%Mo2C/Cg | H2 | 14.2 | 9.6 | 4.3 | 2.8 | 0 | 52.1 | 27.5 | 8.4 | 9.0 | — | — | — | — | |
Interestingly, when both platinum and molybdenum were added to the carbon support to form a Pt–Mo2C/C catalyst, a totally different product distribution was observed. After the reaction at 250 °C under 4.5 MPa CO for 15 min, the formation of 1,2-alkanediols was observed with a yield of 28.7%. For these alkanediol products, the selectivities towards ethylene glycol (EG), 1,2-propanediol (1,2-PD), 1,2-butanediol (1,2-BD) and 1,2-hexanediol (1,2-HD) were 39.4%, 32.0%, 15.0% and 13.6%, respectively (Table 1, entry 4), and these values remained relatively constant when the reaction time was prolonged up to 60 min (ESI, Fig. S2a†). As shown in Scheme S1 (see ESI†), besides the hydrolysis and/or retro-aldol condensation processes, the formation of alkanediols that originated from C–C hydro-cracking and hydrodeoxygenation of glucose, the reaction intermediate, required the participation of hydrogen. Since no gas-phase hydrogen was used, it is reasonable to deduce that hydrogen species were generated in situ in this reaction system. These species then take part in the subsequent reaction over the Pt metal centers, leading to the hydrogenation of the reaction intermediates and thereby the formation of polyols. The hydrogen was not from the cellulose reforming process, as no polyols were obtained when the reaction was conducted under N2 (Table 1, entry 6). Very recently, Schweitzer et al. reported that a Mo2C-supported Pt catalyst exhibited high activity for the WGS reaction at 240 °C.21 It is therefore possible that the hydrogen species produced from the WGS reaction over the Pt–Mo2C/C catalyst were used in the hydrogenation of the cellulose. Indeed, when all other reaction parameters were maintained except for the removal of the cellulose from the reactants, we observed good WGS activity by the Pt–Mo2C/C catalyst with a very high TOF of 3.54 molCO molPt−1 s−1. This value is much higher than that of the Pt/C catalyst under the same reaction conditions (0.04 molCO molPt−1 s−1), and even higher than that of the Pt/Mo2C catalyst (2.19 molCO molPt−1 s−1). The catalysts became deactivated after long term use, but the catalytic activity could be restored through catalyst recarbonization processes. The conversions of glucose and treated cellulose under CO–water at different conditions were also studied. As glucose tends to coke at relatively higher temperatures, the reactions were conducted at temperatures lower than 200 °C. However, at relatively low reaction temperatures, the Pt–Mo2C/C catalyst is not very efficient for the water-gas shift (WGS) reaction. As a result, although glucose is much easier to be converted as compared with cellulose, the less efficient WGS activity makes the overall enhancement less pronounced. The data are listed in Table S3.† The effect of pressure was also investigated which suggests that different to the hydrogen-assisted biomass hydrogenation reaction, the CO-mediated reaction is relatively independent of pressure.
To understand the reaction path, we used (scanning) transmission electron microscopy (S)TEM to characterize the morphology, structure and composition of the composite catalysts. High-resolution TEM (HRTEM) images and statistical analysis indicated that spherical and fcc-structured Pt nanoparticles were well dispersed over the carbon support (3.5 ± 1.0 nm) in the Pt/C catalyst (ESI, Fig. S3a†). In the case of the Mo2C/C catalyst, crystalline β-Mo2C nanoparticles with random orientations and a wide size distribution (20–50 nm) were easily distinguished on the amorphous carbon supports (ESI, Fig. S4†). The Fast Fourier Transform (FFT) of selected single particles could be perfectly indexed by the hexagonal Mo2C phase, indicating their single-crystal nature. The (S)TEM images of the Pt–Mo2C/C catalyst are shown in Fig. 2. Due to the very different atomic numbers of C, Mo and Pt, it is easy to distinguish Pt particles (∼5 nm), Mo2C particles (20–50 nm) and the carbon support from each other by the Z-contrast of high-angle annular dark-field (HAADF)-STEM. As shown in Fig. 2a, small Pt nanoparticles indicated by the brightest regions in the image are uniformly distributed on both the Mo2C nanoparticles (slightly darker regions) and the carbon support (darkest regions). Energy dispersive X-ray spectroscopy (EDX) mapping in Fig. 2b confirms the assignments of different nanoparticles on the carbon support by showing the spatial distributions of Mo and Pt. In addition, the FFT diffractogram of a HRTEM image confirms the coexistence of Pt and Mo2C nanocrystals in the local area (Fig. 2c). Apparently, there are two distinct types of active domains, Pt–C (Pt particles dispersed over carbon support) and Pt–Mo2C (composite of Pt particles–Mo2C particles), which co-exist and are in intimate contact in Pt–Mo2C/C (Fig. 2a and 1f). Considering the different Fermi energies of Mo2C and carbon, the Pt nanoparticles in the two domains should have very different electronic properties.
The different electronic properties of the two types of Pt nanoparticles in the two domains of the Pt–Mo2C/C catalysts were verified by CO temperature programmed desorption (CO-TPD) experiments. The CO adsorbed on Pt nanoparticles with different electronic structures should have different binding energies and therefore different desorption temperatures on the CO-TPD profile. It is clear in Fig. 1d that the adsorption of CO on the Mo2C/C catalysts was rather weak, while a sharp desorption peak on the Pt/C catalysts was observed at around 120 °C. The desorption temperature of the Pt/Mo2C catalyst was much lower, at around 75 °C, indicating remarkably weakened CO binding with the Pt surface. It is not surprising to see that the CO desorption from the Pt–Mo2C/C catalyst produced two peaks at 75 °C and 120 °C, corresponding to CO desorption from the Pt–Mo2C domains and from the Pt–C domains, respectively.
Given the above reactions, TEM characterization results and CO-TPD results, we propose that Pt–Mo2C/C constitutes a unique bifunctional catalyst system (see Fig. 1f), i.e., the WGS reaction occurs on the Pt–Mo2C domains while the subsequent hydrogenation/hydrogenolysis takes place on the Pt–C domains, which have good hydrogenation capabilities. Notably, despite its excellent WGS activity, the Pt/Mo2C catalyst gives rise to a low yield of polyols (<1%) under a CO atmosphere (Table 1, entry 3), implying that it is inactive for the subsequent hydrogenation/hydrogenolysis. To verify this, we tested various catalysts for cellulose transformation reactions using gas-phase hydrogen. As shown in Table 1, entries 7–9 and Fig. 1e, the yield of polyols for Pt/Mo2C was very low under a hydrogen atmosphere, confirming its low hydrogenation activity; on the contrary, both Pt/C and Pt–Mo2C/C catalysts, which have carbon-supported Pt nanoparticles (Pt–C domains), had good selectivity for polyol production. The optimization of reaction parameters needs further research while Fig. S2b–d† provide some preliminary results.
The molecular polarity of CO is larger than H2, so the solubility of CO is higher than that of H2 in polar solvent, e.g., water.22 At the same time, the binding of CO on Pt is much stronger than that of H2. Both may contribute to the excellent catalytic performance of the CO–water–Pt system. The CO adsorbed on the surface is apt to react with the water to form reactive hydrogen species for the subsequent hydrogenation reaction. It should be noted that when these cellulose hydrogenation reactions were conducted under 4.5 MPa hydrogen and 250 °C, the yield of polyols on the Pt–Mo2C/C catalysts was lower than when under a CO atmosphere with similar reaction conditions (Table 1, entry 9 vs. entry 4). This result suggests that in reactions under a CO atmosphere, the WGS reaction over the Pt–Mo2C domain may produce highly active surface hydrogen species in addition to gaseous hydrogen. The surface hydrogen species might migrate onto the adjacent Pt–C domain and accomplish subsequent hydrogenation of the reaction intermediates. These species have better hydrogenation capabilities than gas-phase hydrogen, as indicated by the yield from the hydrogenation reaction of the CO atmosphere cellulose conversion reaction (28.7%) being higher than that from the hydrogen atmosphere reaction (14.2%) (Table 1). Surface hydrogen species were also proposed to be involved in the gold-catalyzed reduction of nitrobenzene by CO and H2O. This reaction has a very good hydrogenation ability, even at room temperature.12a
Our hypothesis that Pt–Mo2C/C is a type of tandem catalyst system, with Pt–Mo2C domains responsible for hydrogen generation and Pt–C domains responsible for subsequent hydrogenation reactions is therefore confirmed. The intimate integration of the two types of domains over the carbon support is important in a tandem catalyst, as verified by a control experiment in which we used a physical mixture of Pt/Mo2C and Pt/C as a catalyst for the cellulose conversion reaction under identical reaction conditions in a CO atmosphere, with a yield of only 2.7% polyols (ESI, Table S1†). This result suggested that an effective coupling of the two reactions, which can be achieved by the hierarchical structure of the Pt–Mo2C/C catalyst (see Fig. 1f), is essential to the overall performance of the catalyst in hydrogen-free transformation of cellulose.
The passivated Pt–Mo2C/C catalyst yielded little hexitol, possibly because it contains molybdenum oxide, which facilitated the C–C bond cleavage reaction.20 We investigated the unpassivated Pt–Mo2C/C catalyst in the cellulose transformation reaction with CO and H2O as the hydrogen source. Specifically, freshly synthesized Pt–Mo2C/C from a TPR process was added directly to the aqueous reaction medium under the protection of N2. Interestingly, both a higher total yield of polyols (42.1%) and a higher selectivity to hexitols (∼60%) were achieved in this way (Table 1, entry 5). These results suggested that the surface properties of the catalyst indeed have a major impact on the behavior of the catalyst and that pristine catalysts possess higher glucose hydrogenation activity, but lower C–C bond cleavage activity (ESI, Scheme S1†). To understand the influence of the solid nature of cellulose, experiments with a much simpler substrate, glucose, were conducted under milder reaction conditions (Table S3–S5†), but less hexitols were obtained than those with cellulose substrate. This was probably due to the fact that the reaction rate of the WGS reaction when using Pt–Mo2C/C is low at low temperatures, which makes the successive hydrogenation less efficient. As a result, the catalytic performance is not as good as expected. Our next goal is to find a catalyst with very good low temperature WGS reaction activity and couple it with the biomass conversion reaction efficiently.
Conclusions
We developed a novel and alternative strategy for the catalytic transformation of cellulose into polyols with low-cost and readily available CO and H2O as the hydrogen source. We fabricated a new tandem Pt–Mo2C/C catalyst in which the Pt–Mo2C domains are responsible for the formation of active hydrogen species from the water-gas shift reaction, while the Pt–C domains catalyze the subsequent hydrogenation/hydrogenolysis reactions. Although more work, such as further optimization of the reaction conditions etc., is required, our current results provide not only a new base for the design of next-generation biomass conversion catalysts, but also a new strategy for the development of green and sustainable chemistry in general.Acknowledgements
This work received financial support from the Natural Science Foundation of China (21173009, 21222306) and two 973 Projects (2011CB201402, 2013CB933100). Y.H. thanks King Abdullah University for baseline funds.Notes and references
- (a) M. Stocker, Angew. Chem., Int. Ed., 2008, 47, 9200 CrossRef CAS PubMed; (b) G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed; (c) J. R. Regalbuto, Science, 2009, 325, 822 CrossRef PubMed; (d) P. Mäki-Arvela, B. Holmbom, T. Salmi and D. Y. Murzin, Catal. Rev. Sci. Eng., 2007, 49, 197 CrossRef.
- (a) T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090 CrossRef CAS PubMed; (b) C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588 RSC; (c) R. Rinaldi and F. Schuth, ChemSusChem, 2009, 2, 1096 CrossRef CAS PubMed; (d) S. Van De Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82 CrossRef CAS.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446 CrossRef CAS PubMed.
- D. Mohan, C. U. Pittman, Jr and P. H. Steele, Energy Fuels, 2006, 20, 848 CrossRef CAS.
- (a) G. Wen, Y. Xu, Z. Xu and Z. Tian, Catal. Commun., 2010, 11, 522 CrossRef CAS PubMed; (b) R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964 CrossRef CAS PubMed.
- (a) H. de Lasa, E. Salaices, J. Mazumder and R. Lucky, Chem. Rev., 2011, 111, 5404 CrossRef CAS PubMed; (b) R. M. Navarro, M. C. Sánchez-Sánchez, M. C. Alvarez-Galvan, F. d. Valle and J. L. G. Fierro, Energy Environ. Sci., 2009, 2, 35 RSC.
- (a) J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164 CrossRef CAS PubMed; (b) J. Z. Zhang, X. Liu, M. Sun, X. H. Ma and Y. Han, ACS Catal., 2012, 2, 1698 CrossRef CAS; (c) J. Z. Zhang, X. Liu, M. N. Hedhili, Y. H. Zhu and Y. Han, ChemCatChem, 2011, 3, 1294 CrossRef CAS.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161 CrossRef CAS PubMed.
- N. Yan, C. Zhao, C. Luo, P. J. Dyson, H. Liu and Y. Kou, J. Am. Chem. Soc., 2006, 128, 8714 CrossRef CAS PubMed.
- (a) C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636 CrossRef CAS PubMed; (b) N. Ji, T. Zhang, M. Zheng, A. Wang, H. Wang, X. Wang and J. G. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510 CrossRef CAS PubMed; (c) Y. Liu, C. Luo and H. Liu, Angew. Chem., Int. Ed., 2012, 51, 3249 CrossRef CAS PubMed; (d) M. Benoit, A. Rodrigues, Q. Zhang, E. Fourré, K. De Oliveira Vigier, J.-M. Tatibouët and F. Jérôme, Angew. Chem., Int. Ed., 2011, 50, 8964 CrossRef CAS PubMed; (e) J. Hilgert, N. Meine, R. Rinaldi and F. Schuth, Energy Environ. Sci., 2013, 6, 92 RSC; (f) Z. Tai, J. Zhang, A. Wang, M. Zheng and T. Zhang, Chem. Commun., 2012, 48, 7052 RSC; (g) S. M. Hick, C. Griebel, D. T. Restrepo, J. H. Truitt, E. J. Buker, C. Bylda and R. G. Blair, Green Chem., 2010, 12, 468 RSC; (h) X. C. Wang, L. Q. Meng, F. Wu, Y. J. Jiang, L. Wang and X. D. Mu, Green Chem., 2012, 14, 758 RSC; (i) R. Palkovits, K. Tajvidi, J. Procelewska, R. Rinaldi and A. Ruppert, Green Chem., 2010, 12, 972 RSC; (j) J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Chem. Commun., 2011, 47, 5590 RSC; (k) J. Geboers, S. Van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577 RSC; (l) M. Liu, W. P. Deng, Q. H. Zhang, Y. L. Wang and Y. Wang, Chem. Commun., 2011, 47, 9717 RSC; (m) W. P. Deng, X. S. Tan, W. H. Fang, Q. H. Zhang and Y. Wang, Catal. Lett., 2009, 133, 167 CrossRef CAS PubMed; (n) R. Palkovits, K. Tajvidi, A. M. Ruppert and J. Procelewska, Chem. Commun., 2011, 47, 576 RSC.
- (a) J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979 CrossRef CAS PubMed; (b) R. Rinaldi, R. Palkovits and F. Schueth, Angew. Chem., Int. Ed., 2008, 47, 8047 CrossRef CAS PubMed; (c) B. Kim, J. Jeong, D. Lee, S. Kim, H.-J. Yoon, Y.-S. Lee and J. K. Cho, Green Chem., 2011, 13, 1503 RSC.
- (a) L. He, L. C. Wang, H. Sun, J. Ni, Y. Cao, H. Y. He and K. N. Ean, Angew. Chem., Int. Ed., 2009, 48, 9538 CrossRef CAS PubMed; (b) L. He, F. J. Yu, X. B. Lou, Y. Cao, H. Y. He and K. N. Fan, Chem. Commun., 2010, 46, 1553 RSC; (c) J. Ni, L. He, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Chem. Commun., 2011, 47, 812 RSC.
- L. Ramqvist, K. Hamrin, G. Johansso, A. Fahlman and C. Nordling, J. Phys. Chem. Solids, 1969, 30, 1835 CrossRef CAS.
- W. A. Brainard and D. R. Wheeler, J. Vac. Sci. Technol., 1978, 15, 1800 CrossRef CAS.
- N. Perret, X. Wang, L. Delannoy, C. Potvin, C. Louis and M. A. Keane, J. Catal., 2012, 286, 172 CrossRef CAS PubMed.
- D. J. Moon and J. W. Ryu, Catal. Lett., 2004, 92, 17 CrossRef CAS.
- D. R. Butcher, M. E. Grass, Z. Zeng, F. Aksoy, H. Bluhm, W. X. Li, B. S. Mun, G. A. Somorjai and Z. Liu, J. Am. Chem. Soc., 2011, 133, 20319 CrossRef CAS PubMed.
- K. Segawa and W. K. Hall, J. Catal., 1982, 76, 133 CrossRef CAS.
- (a) Y. Roman-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954 CrossRef CAS PubMed; (b) Y. Roman-Leshkov and M. E. Davis, ACS Catal., 2011, 1, 1566 CrossRef CAS.
- O. Munoz-Muniz, M. Quintanar-Audelo and E. Juaristi, J. Org. Chem., 2003, 68, 1622 CrossRef CAS PubMed.
- N. M. Schweitzer, J. A. Schaidle, O. K. Ezekoye, X. Pan, S. Linic and L. T. Thompson, J. Am. Chem. Soc., 2011, 133, 2378 CrossRef CAS PubMed.
- Lange's Handbook of Chemistry, 15th edn, McGraw Hill, New York, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ee41945b |
| This journal is © The Royal Society of Chemistry 2014 |