 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the mechanism of the hydroboration of alkenes by amine–boranes catalysed by [Rh(xantphos)]+†
Heather C.
Johnson
a,
Rebecca
Torry-Harris
a,
Laura
Ortega
b,
Robin
Theron
c,
J. Scott
McIndoe
c and
Andrew S.
Weller
*a
aDepartment of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: andrew.weller@chem.ox.ac.uk
bVisiting Student from Departamento de Química Inorgańica, Instituto de Investigaciones Químicas (IIQ), Universidad de Sevilla, Consejo Superior de Investigaciones Científicas, Avda. Ameŕico Vespucio 49, 41092 Sevilla, Spain
cDepartment of Chemistry, University of Victoria, P.O. Box 3065, Victoria, BC V8W 3V6, Canada
First published on 6th June 2014
Abstract
The [Rh(xantphos)]+ fragment acts as an effective catalyst for the hydroboration of the alkene TBE (tert-butyl ethene) using the amine–borane H3B·NMe3 at low (0.5 mol%) catalyst loadings to give the linear product. Investigations into the mechanism using the initial rate method and labelling studies show that reductive elimination of the linear hydroboration product is likely the rate-limiting step at the early stages of catalysis, and that alkene and borane activation (insertion into a Rh–H bond and B–H oxidative addition) are reversible. The resting state of the system has also been probed using electrospray ionization mass spectrometry (ESI-MS) using the pressurised sample infusion (PSI) technique. This system is not as effective for hydroboration of other alkenes such as 1-hexene, or using phosphine borane H3B·PCy3, with decomposition or P–B bond cleavage occurring respectively.
Introduction
Hydroboration, the addition of a B–H bond across an unsaturated C–C bond, is a versatile methodology that affords organoboranes, from which subsequent functionalisation leads to products of use in organic synthesis.1–4 Non-metal catalysed hydroboration generally yields the anti-Markovnikov product, whereas transition metal catalysts enable control over the regioselectivity of hydroboration. Such selectivity (i.e. linear versus branched products) has been shown to vary with different catalysts, alkenes and even reaction conditions.5–8 Historically metal-catalysed hydroborations have used three-coordinate boron substrates such as catechol (HBCat) or pinacol borane.2,3 By contrast four-coordinate amine–boranes (prototypically H3B·NMe3) have traditionally been used in uncatalysed hydroboration where N–B cleavage is proposed to afford a reactive trivalent BH3 molecule,9 although iodine-induced hydroboration is proposed to operate via an intermediate that retains the B–N bond.10 Amine–boranes have, instead, received much recent attention due to their potential as hydrogen storage systems and as precursors to oligomeric or polymeric B–N materials via dehydrocoupling;11 and we,12,13 alongside others,9,11 have been exploring the role of the metal catalyst in these processes. Recognising that B–H oxidative cleavage from a bound sigma complex to form a metal boryl hydride (Scheme 1a) is closely related to the same mode of activation of a B–H bond at a metal in hydroboration (Scheme 1b), we reported in 2011 that the addition of the alkene tert-butylethene (TBE) to the sigma amine–borane complex [Rh(PiBu2tBu)2(η2-H3B·NMe3)][BArF4] resulted in the formation of the linear hydroboration product [Rh(PiBu2tBu)2(η2-H2B(CH2CH2tBu)·NMe3)][BArF4]14 [ArF = 3,5-(CF3)2C6H4]. The precursor complex [Rh(PiBu2tBu)2][BArF4] also slowly (94 h, 5 mol%) catalysed this process to form free H2B(CH2CH2tBu)·NMe3 (I), Scheme 1c.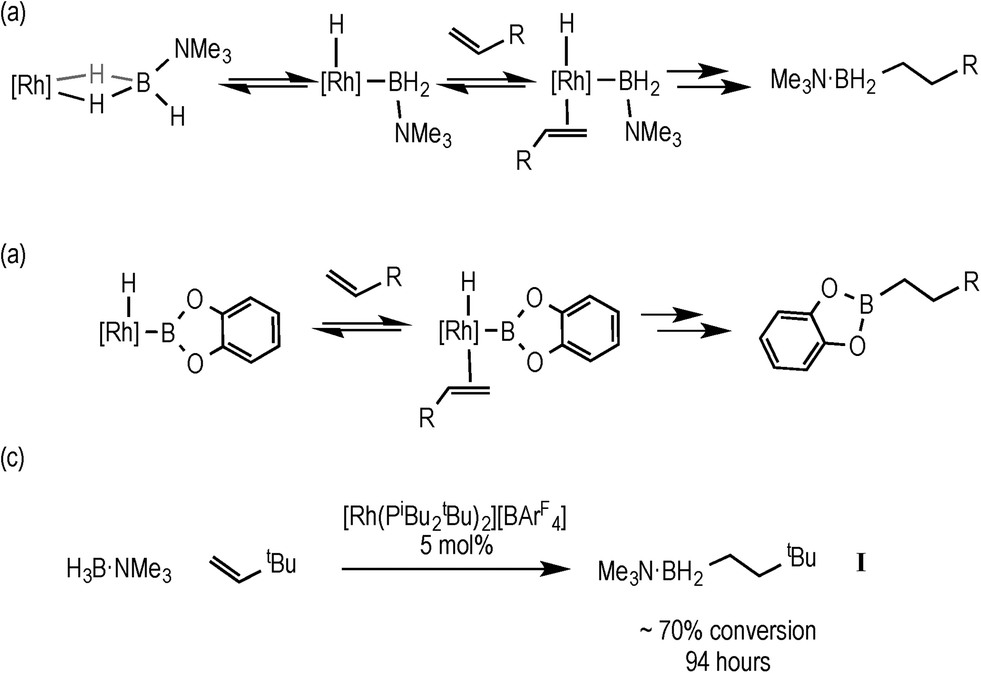 | ||
| Scheme 1 Rh-catalysed hydroboration using amine–borane (a) and catechol borane (b); hydroboration of TBE using H3B·NMe3, (c). | ||
Kinetic experiments allowed for a mechanism to be proposed in which the hydroborated product inhibited catalytic turnover and reductive elimination of the product was also suggested to be slow.15 Independently, in 2012, a similar methodology using N-heterocyclic carbene–boranes and chiral Rh-based catalysts was reported for intramolecular hydroborations of alkenes.16 Very recently we briefly communicated that by using a [Rh(κ2P,P-xantphos)]+ based catalyst,17 TBE can be hydroborated to give I. In the absence of this alkene, dehydrogenative homocoupling of the borane occurs (see Scheme 4), a process suggested to occur via the B–H activated intermediate that is no longer intercepted by coordination of alkene.18 We now report in detail on this hydroboration, including kinetic data that support a proposed mechanism, as well as assessing the scope of this catalyst with regard to other alkenes and phosphine–boranes.
Results and discussion
Preliminary stoichiometric and catalytic studies
Addition of excess TBE to the Rh(III) sigma–borane complex [Rh(κ3P,O,P-xantphos)(H)2(η1-H3B·NMe3)][BArF4] 1 resulted in the rapid formation (less than 5 minutes) of the Rh(I) complex [Rh(κ2P,P-xantphos)(η2-H2B(CH2CH2tBu)·NMe3)][BArF4] (2) as the sole metal-containing product (Scheme 2), presumably by initial hydrogenation of one equivalent of alkene to form a Rh(I) species, followed by hydroboration of another equivalent. The solid-state structure and NMR spectroscopic data for 2 have previously been communicated.18 In a similar manner, addition of trimethylvinyl silane to 1 gives the equivalent complex 3, [Rh(κ2P,P-xantphos)(η2-H2B(CH2CH2SiMe3)·NMe3)][BArF4], in which H2B(CH2CH2SiMe3)·NMe3 (II) is bound to the metal centre.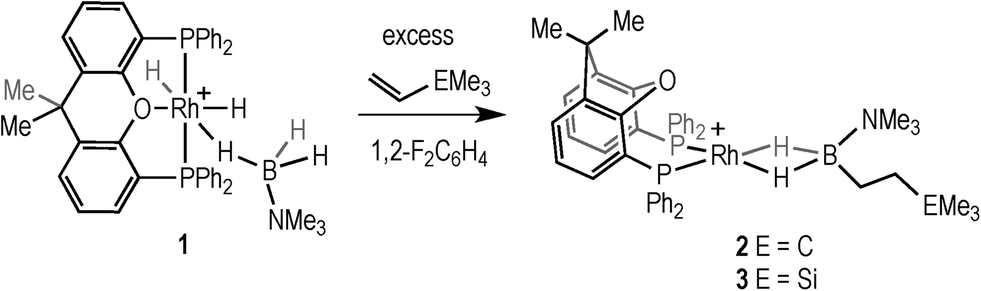 | ||
| Scheme 2 Formation of 2 (E = C) and 3 (E = Si). [BArF4]− anions not shown. | ||
Complex 3 was characterised by NMR spectroscopy, ESI-MS (electrospray ionisation mass spectrometry) and microanalysis, which together show similar analytical data to 2 and closely related [Rh(PiBu2tBu)2(η2-H2B(CH2CH2tBu)·NMe3)][BArF4].14 The alkyl borane binds to the metal centre through two sigma Rh–H–B interactions, evident by single 11B quadrupolar-broadened signal at δ −6.54 in the 1H NMR spectrum of relative integral 2H, which collapses to an overlapping doublet of doublets (virtual triplet) on decoupling to 11B [J(RhH) = 36 Hz, J(PtransH) = 36 Hz]. Two, relative integral 2H, multiplets were observed at δ 1.17 and δ 0.78 for the CH2 groups, indicating that the anti-Markovnikov (i.e. linear) product of hydroboration is bound to the metal centre. A 29Si-1H HMBC NMR experiment showed a correlation between silicon [δ(29Si) 2.1] and the alkyl protons at δ 1.17, assigning these to those α to Si. The xantphos methyl groups are observed as two separate environments (δ 1.73 and δ 1.67). In the 11B NMR spectrum a broad resonance is observed at δ 37, typical for η2-coordination of an amine–borane to a RhI centre,19,20 which has shifted 45.7 ppm downfield from that in 1 (δ −8.7).18 Similar changes in 11B chemical shift have been noted in related systems on moving between Rh(I) and Rh(III) oxidation states.19,21 The 31P{1H} NMR spectrum shows a single environment δ 26.7 [d, J(RhP) = 182 Hz]. The solid-state structure of complex 3 supports the solution data (Fig. 1), in particular a close Rh⋯B distance of 2.179(7) Å, which is the same within error to that found in 2, 2.162(5) Å,18 and the formation of the linear hydroboration product. Complexes such as 2 and 3 are valence isoelectronic analogs of sigma alkane complexes,22–24 while related alkyl sigma amine–borane complexes have previously been prepared.25
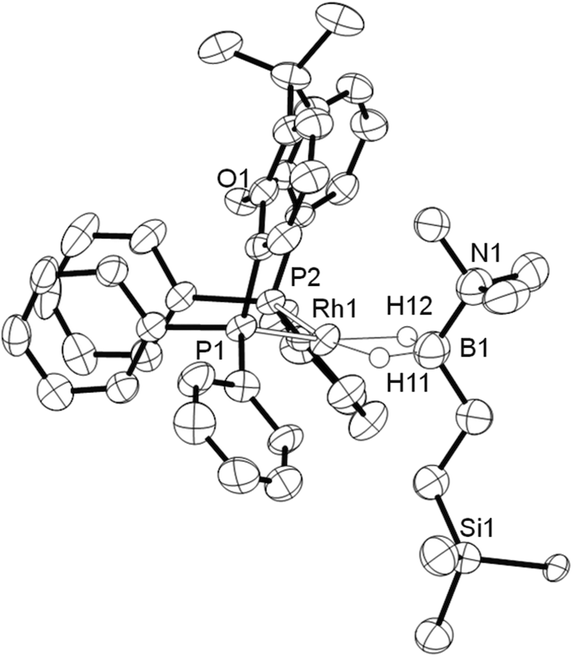 | ||
| Fig. 1 Solid-state structure of the cationic portion of 3. Displacement ellipsoids are drawn at the 50% probability level. [BArF4]− anion and all carbon-bound H atoms are omitted for clarity. Only the major component of the disordered NMe3 and SiMe3 groups are shown. Selected bond lengths (Å) and angles (°): Rh1–P1, 2.2398(18); Rh1–P2, 2.2670(17); Rh1–O1, 3.2342(73); Rh1–B1, 2.179(7); B1–N1, 1.603(4); P1–Rh1–P2, 98.23(6). | ||
With complexes 2 and 3 in hand the catalytic hydroboration of TBE with H3B·NMe3 was explored using these as precatalysts (Scheme 3). As previously reported,18 complex 2 (5 mol%) catalyses the complete conversion to H2B(CH2CH2tBu)·NMe3 (I) from TBE and H3B·NMe3 within 3 hours. The catalysis was conducted with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of alkene:H3B·NMe3 as the {Rh(xantphos)}+ fragment has been reported to promote the slow dehydrogenative homocoupling of H3B·NMe3 to form [Rh(κ2P,P-xantphos)(η2-H4B2·2NMe3)][BArF4] (4) alongside 1 (Scheme 4),18 and a two-fold excess of alkene prevents the formation of 4 in detectable quantities (vide infra). During catalysis, complex 2 was the only observed resting state by 1H and 31P{1H} NMR spectroscopy.26 The 1H NMR spectrum of isolated I confirms anti-Markovnikov regioselectivity, with two, integral 2H, multiplets at δ 1.42 and δ 0.55 assigned to the methylene groups. The 11B NMR spectrum shows a triplet at δ −0.83 [J(BH) = 96 Hz].14
:1 ratio of alkene:H3B·NMe3 as the {Rh(xantphos)}+ fragment has been reported to promote the slow dehydrogenative homocoupling of H3B·NMe3 to form [Rh(κ2P,P-xantphos)(η2-H4B2·2NMe3)][BArF4] (4) alongside 1 (Scheme 4),18 and a two-fold excess of alkene prevents the formation of 4 in detectable quantities (vide infra). During catalysis, complex 2 was the only observed resting state by 1H and 31P{1H} NMR spectroscopy.26 The 1H NMR spectrum of isolated I confirms anti-Markovnikov regioselectivity, with two, integral 2H, multiplets at δ 1.42 and δ 0.55 assigned to the methylene groups. The 11B NMR spectrum shows a triplet at δ −0.83 [J(BH) = 96 Hz].14
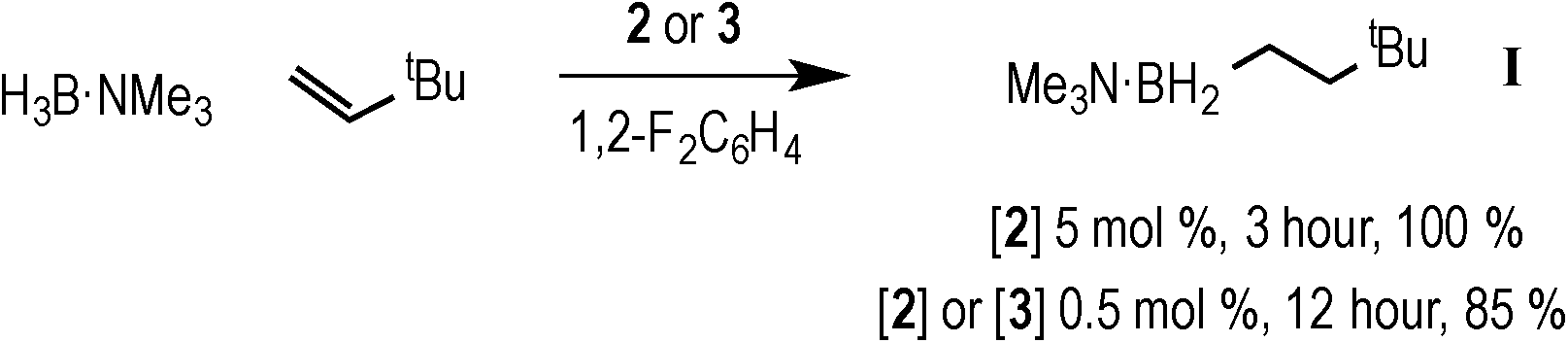 | ||
| Scheme 3 Rhodium-catalysed hydroboration of TBE by H3B·NMe3. | ||
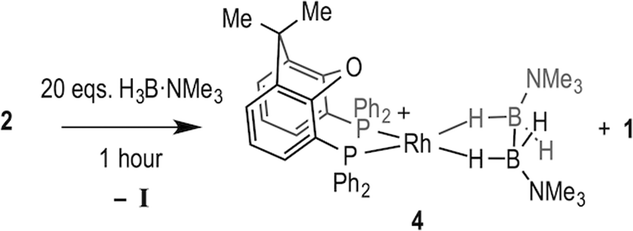 | ||
| Scheme 4 Formation of complex 4 from addition of excess H3B·NMe3 to 2. [BArF4]− anions not shown. | ||
Kinetic studies
Given the promising rate of hydroboration of TBE with catalyst 2 to afford I, the catalyst loading was reduced to 0.5 mol%, relative to H3B·NMe3. Under these conditions ([H3B·NMe3] = 0.19 M, [TBE] = 0.38 M, 1,2-F2C6H4 solvent), consumption of H3B·NMe3 to yield I proceeded to 85% completion after 12 hours as monitored by 11B NMR spectroscopy, with the balance being made by unreacted H3B·NMe3. Longer reaction times did not result in further reaction, suggesting either product inhibition and/or catalyst decomposition. When starting from precatalyst 3 a very similar overall temporal profile was observed, suggesting the identity of the initially bound amine–borane (I or II) does not affect the overall rate of catalysis.The potential for product inhibition,14 and the parallel homocoupling reaction with excess H3B·NMe3, suggested that the method of initial rates was most appropriate to probe the reaction orders with respect to substrates and the catalyst.27 After 300 s at 5 mol% loading of 3ca. 50% substrate conversion had occurred, while at 0.5 mol% this was now only ca. 10% conversion, making the lower loading suitable for study by the initial rate method. We further chose to study catalyst 3 as this would also give additional information as to the evolution of the likely resting states. Table 1 presents the data from this study, and Fig. 2 presents some of these data in graphical format.
| Entry | [3] (10−4 M) | [H3B·NMe3] (M) | [TBE] (M) | Initial ratea (10−5 M s−1) |
|---|---|---|---|---|
| a Calculated from the pseudo zero-order region of the temporal evolution of I as measured by 11B NMR spectroscopy over the first 300 s of catalysis. b With an additional 70 equiv. I at the start of catalysis. c Using D3B·NMe3 instead of H3B·NMe3. | ||||
| 1 | 9.5 | 0.19 | 0.38 | 6.81 ± 0.12 |
| 2 | 9.5 | 0.19 | 0.19 | 3.64 ± 0.27 |
| 3 | 9.5 | 0.19 | 0.76 | 12.98 ± 0.38 |
| 4 | 19.0 | 0.19 | 0.38 | 13.25 ± 0.56 |
| 5 | 9.5 | 0.38 | 0.19 | 7.41 ± 0.53 |
| 6 | 9.5 | 0.76 | 0.19 | 6.33 ± 0.40 |
| 7b | 9.5 | 0.19 | 0.38 | 2.07 ± 0.11 |
| 8 | [2] 9.5 | 0.19 | 0.38 | 7.44 ± 0.64 |
| 9 | 9.5 | 0.19c | 0.38 | 5.09 ± 0.07 |
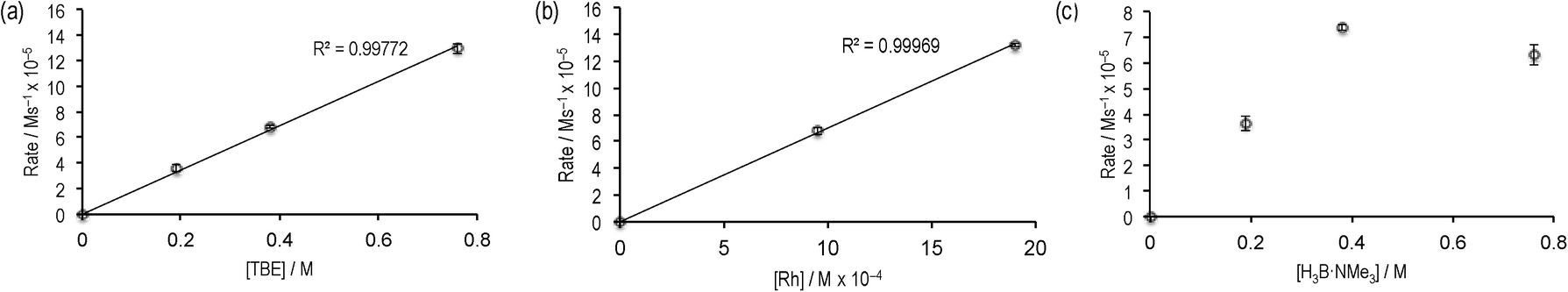 | ||
| Fig. 2 Initial rate experiments: (a) variation of [TBE]; (b) variation of [3]; (c) variation of H3B·NMe3. See Table 1 for more details. | ||
Comparison of entries 1, 2 and 3 (Fig. 2a) show that the reaction is essentially first order in [TBE]. Entries 1 and 4 demonstrate a first order relationship in catalyst 3 (Fig. 2b). Entries 2, 5 and 6 show that increasing the concentration of [H3B·NMe3] moves from an approximate first order relationship to an inhibition of catalysis at higher concentrations of amine–borane (Fig. 2c), we presume as homocoupling to form 4 becomes competitive. In complex 4, the diborane(4) is relatively strongly bound to the metal centre, remaining intact even with the addition of MeCN,18 and thus is unlikely to be as active in catalysis. Indeed, use of 4 as a catalyst (0.5 mol%) resulted in reduced turnover. Addition of excess product I (~70 equivalents, entry 7) results in a significant slowing of the initial rate, consistent with strong product inhibition, as observed to a lesser degree with the [Rh(PiBu2tBu)2][BArF4] system.14 Catalyst 3 and catalyst 2 operated at the same initial rate, within error (entries 1 and 8), suggesting that the identity of the bound primary borane (i.e.I or II) does not influence initial rate of turnover.
Hydroboration of TBE and H3B·NMe3 catalysed using 3 enables more information to be gleaned about possible resting states. At 0.5 mol% loading, the catalyst concentration is too low to be observable by NMR spectroscopy under the conditions used. However, at 5 mol% loading the rhodium-containing species can be probed by 1H and 31P{1H} NMR spectroscopy. The diagnostic, broad, hydride signals for 2 and 3 appear at similar chemical shifts in 1,2-F2C6H4 solvent [δ −6.8518 and δ −6.54 respectively]. In the early stages of catalysis (~20% conversion), the 1H NMR spectrum shows a mixture of 2 and 3, evident by broad overlapping hydride peaks. As catalysis progresses, this broad overlapping resonance sharpens and 2 becomes the dominant species demonstrating that I displaces II in the resting state during catalysis. Under the conditions of excess H3B·NMe3 (cf. entry 6), using 5 mol% 3 to enable monitoring by 1H NMR spectroscopy, complex 4 grows in over time, whereas under conditions of excess TBE it is not observed. This is consistent with the kinetic data that suggest removal from the system of active catalyst at high [H3B·NMe3], leading to inhibition.
The change in resting state from 3 to 2 has also been probed using electrospray ionization mass spectrometry (ESI-MS) using the pressurised sample infusion (PSI) technique.28–30 The particular advantage of this technique is that it allows for very high data density over a wide dynamic range, and is thus ideal for analysing evolving mixtures during catalysis. Fig. 3 shows the temporal profile of the catalysis using 3. This experiment was run at 15 mol%, which was determined to be the best conditions for the optimal (low) concentration necessary for PSI-ESI-MS. Immediately at the start of catalysis the resting state moves from 3 to 2, consistent with the NMR experiments. These ESI-MS experiments also reveal the presence, at early stages of the reaction of three other species. The first is identified as [Rh(xantphos)(H3B·NMe3)]+, (m/z = 754.24; calc. 754.20), although we cannot comment on the precise structure: it could be a Rh(I) sigma-bound amine–borane complex, or a Rh(III) B–H activated hydrido-boryl. Both structural forms have precedent19,31 and are likely to be in equilibrium with one another.32 Indeed both have been calculated to be accessible, but thermodynamically unfavoured, compared with 2.18 [Rh(xantphos)(H2)]+ (m/z = 683.15; calc. 683.11) and [1]+ are also observed, which we suggest both come from a small amount of [1]+ formed parallel with 4 during catalysis (Scheme 4). That we do not observe any of these species by 1H NMR spectroscopy (hydride region) suggests that ESI-MS is particularly sensitive to their observation. These species decay at a very similar rate to [3]+, which suggests that the build-up of I during catalysis pushes any equilibria operating to favour of 2. This observation is also consistent with product inhibition from initial rate experiments.
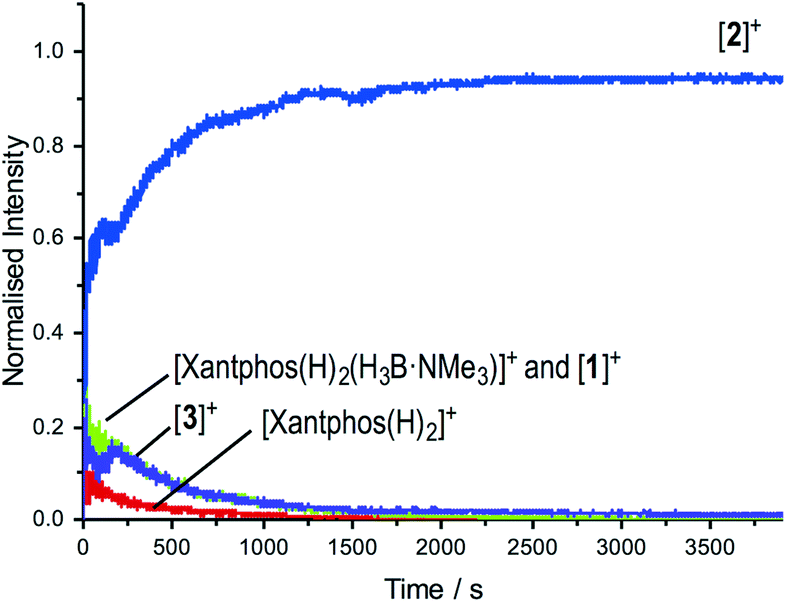 | ||
| Fig. 3 ESI-MS under PSI conditions29 of the reaction of TBE with H3B·NMe3 catalysed by 3. Conditions: H3B·NMe3, 0.006 M, TBE 0.013 M; [3] 0.001 M, 1,2-F2C6H4. Under these conditions of concentration and experiment catalysis proceeded to 80% conversion. | ||
Labelling studies
Complex 3 (and 2) are initially produced under conditions of excess alkene (Scheme 2), suggesting that the alkene does not bind competitively with II (or I), while the dependence of the rate law upon both [TBE] and [H3B·NMe3] indicates that irreversible B–H oxidative addition prior to alkene coordination is not rate-determining. The potential for reversibility of the binding of both H3B·NMe3 and TBE to the metal centre was further probed using D3B·NMe3 instead of H3B·NMe3 during catalysis. Initial rate experiments (Table 1, entry 9) showed a KIE of 1.34 ± 0.04, consistent with irreversible B–H activation not being rate determining. However, due to the H/D exchange observed between the amine–borane and alkene (vide infra) interpretation of the absolute magnitude of this measurement should be treated with a degree of caution.After 1 hour of catalysis under conditions of excess alkene (28% conversion, Scheme 5) 2H NMR spectroscopy showed incorporation of deuterium into the internal position of the free, unreacted, alkene (δ 5.89), while the corresponding signal in the 1H NMR spectrum decreased by ca. 25% relative to the other alkene signals at δ 4.99 and 4.89. This demonstrates that H/D exchange occurs only occurs at the internal alkene proton. H/D exchange in free amine–borane was evidenced by the 11B NMR spectrum that at early stages of catalysis showed a broad peak corresponding to D3B·NMe3 and evolved with time to show significant signs of B–H coupling.33 The final product d-I showed no H/D exchange α- to the borane, and ~40% H/D exchange at the β position (i.e. 60% D).
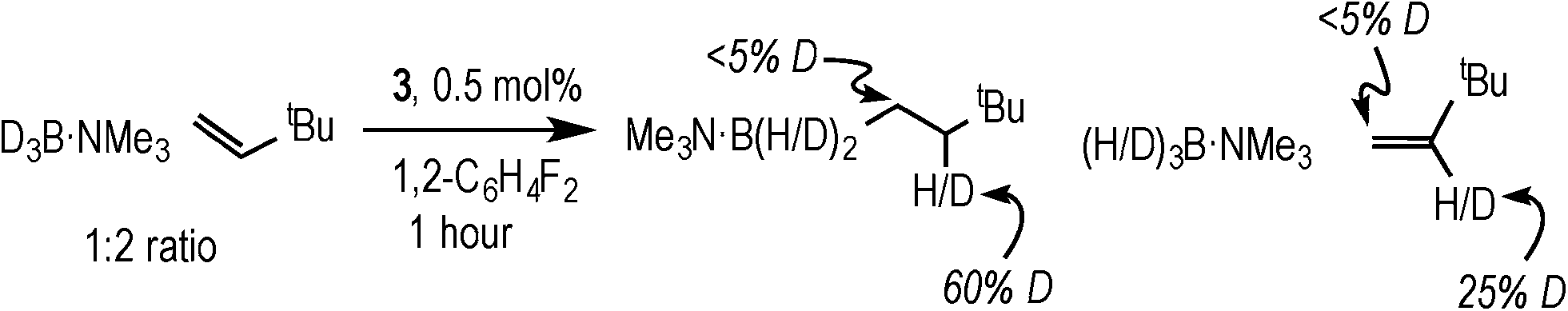 | ||
| Scheme 5 The products observed after 1 hour of catalysis using D3B·NMe3. Conditions: [D3B·NMe3] = 0.19 M, [TBE] = 0.38 M, 1,2-F2C6H4, 0.5 mol% 3. | ||
These data suggest that coordination of H3B·NMe3, B–H activation, coordination and insertion of the alkene into the Rh–H bond are all reversible, to ultimately give the linear product. Moreover the lack of H/D exchange in the final product at the α-position, and a similar lack of exchange in the terminal positions of the free alkene, suggests that insertion to form the branched product is not occurring. We also suggest that hydride migration to the alkene, rather than boryl migration to form an intermediate such as F (Scheme 6), is by far dominant.34 Intermediates such as F have been postulated in dehydrogenative borylation reactions,5,8,35,36 the products of which are not observed here. Although we cannot fully discount that boryl migration from F is reversible but the barrier to reductive elimination from F is high, we consider that this scenario is less likely based upon literature precedent.6–8,35
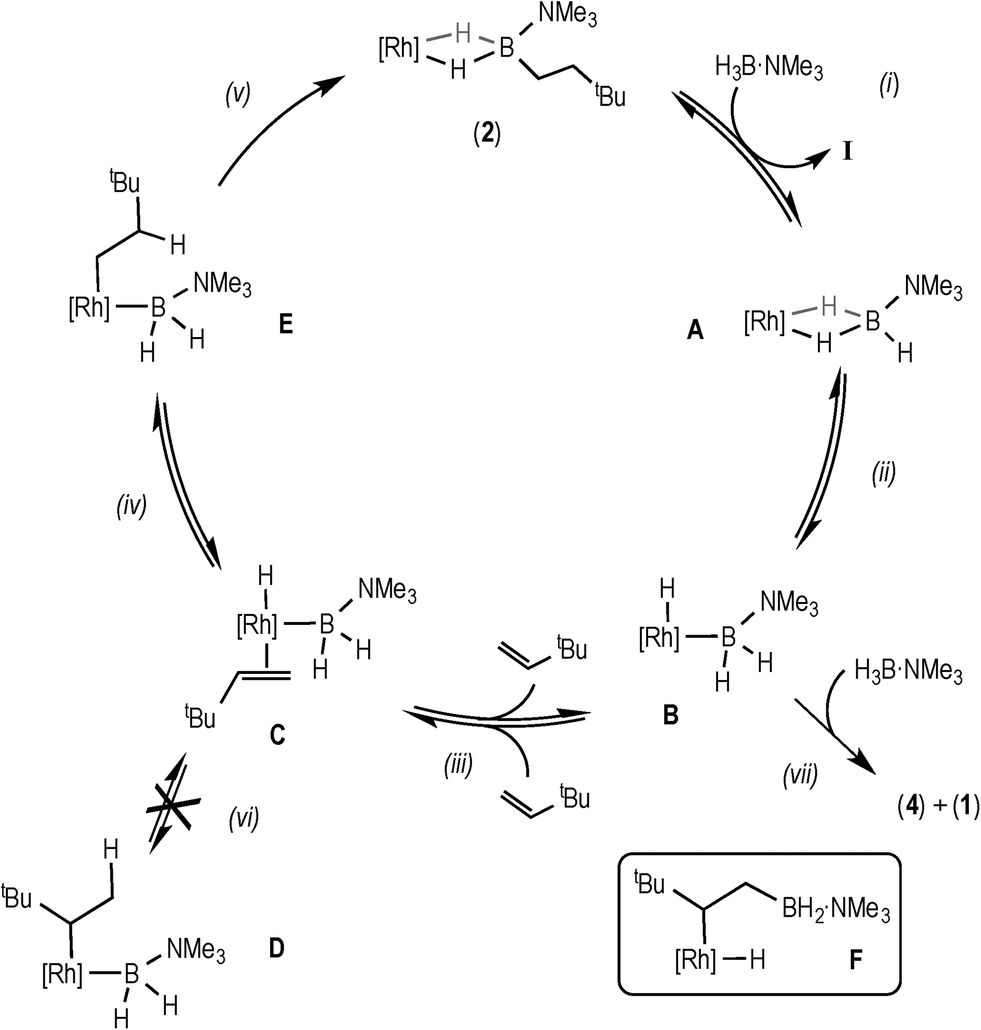 | ||
| Scheme 6 Proposed mechanism for the catalytic hydroboration using data from the early phase of catalysis. [Rh] = [Rh(xantphos)]+. | ||
Bringing these data and observations together leads us to propose the catalytic pathway shown in Scheme 6 for the hydroboration of TBE using H3B·NMe3 and 3. This pathway is similar to that reported for using the [Rh(PiBu2iPr)2]+ catalyst system,14 as well as late transition metal hydroboration systems that use, for example, HBCat.3,5,6,8
The elementary steps in this cycle are thus: complex 2 does not react with TBE but undergoes reversible B–H activation with H3B·NMe3, (i) and (ii), as shown by H/D exchange into free D3B·NMe3 during the early phases of catalysis. Monitoring by ESI-MS shows a species consistent with A or B (m/z = 754.24) before 2 becomes the only species observed. TBE binding and insertion into Rh–H is reversible, (iii), as demonstrated by H/D exchange into the free alkene during catalysis. No branched product is observed37 and no H/D exchange at the α-position of the linear product is measured, showing that insertion from C to form D (vi) is neither kinetically competent nor reversible. Insertion from C to give the linear intermediate E is reversible (iv), as there is significant (40%) H/D exchange at the β-position in the final product, as well as into the free alkene when D3B·NMe3 is used, that suggests that β-H-elimination from E occurs. Overall these H/D labelling experiments suggest that reductive elimination (v) is the turnover-limiting step during the early stages of catalysis. As reductive elimination would be expected to have a small (close to unity) KIE, the modest measured value might reflect a system at equilibrium before the turnover limiting step (as postulated), i.e. an equilibrium isotope effect.38
Hydroboration of alkenes other than TBE
Under stoichiometric conditions the hydroboration of alkenes other than TBE was explored. With the hindered alkene 2,3-dimethyl-2-butene (2 equiv.) no evidence of hydroboration using 1/H3B·NMe3 was observed, with 1 remaining the major organometallic species in solution over an hour. After 24 hours, a 2:1 mixture of 1 and 4 were present, suggesting that the B–B homocoupling was occurring. Cyclohexene behaved similarly, as previously reported, with no hydroboration observed; instead it promotes loss of H2 from 1, driving the formation of 4.18 To probe the possibility of non-productive coordination of cyclohexene at the metal centre, a mixture of D3B·NMe3 (0.19 M), cyclohexene (0.38 M) and 3 (5 mol%, relative to D3B·NMe3) was monitored by 2H NMR spectroscopy. This showed, as well as deuterium incorporation into signals for cyclohexane that arise from deuteration of 1 (δ 1.58 and δ 1.43), incorporation of deuterium into the alkene signal (δ 5.89) after 90 minutes (Scheme 7). These data suggest that reversible alkene coordination and deuteride insertion can occur to give an intermediate similar to E; while the lack of hydroborated product suggests that the reductive elimination, as proposed to be the rate determining step for catalysis with TBE, is slow compared with overall dehydrogenative homocoupling from an intermediate B to form 4. This is presumably related to the relative rates of reductive C–B coupling of primary and secondary alkyl–boryls, which in turn is likely related to barriers to re-orientation of the sp3 alkyl and boryl groups prior to reductive coupling,39 which is expected to be greater for locally bulkier substituents. Thus TBE undergoes hydroboration, while cyclohexene does not.
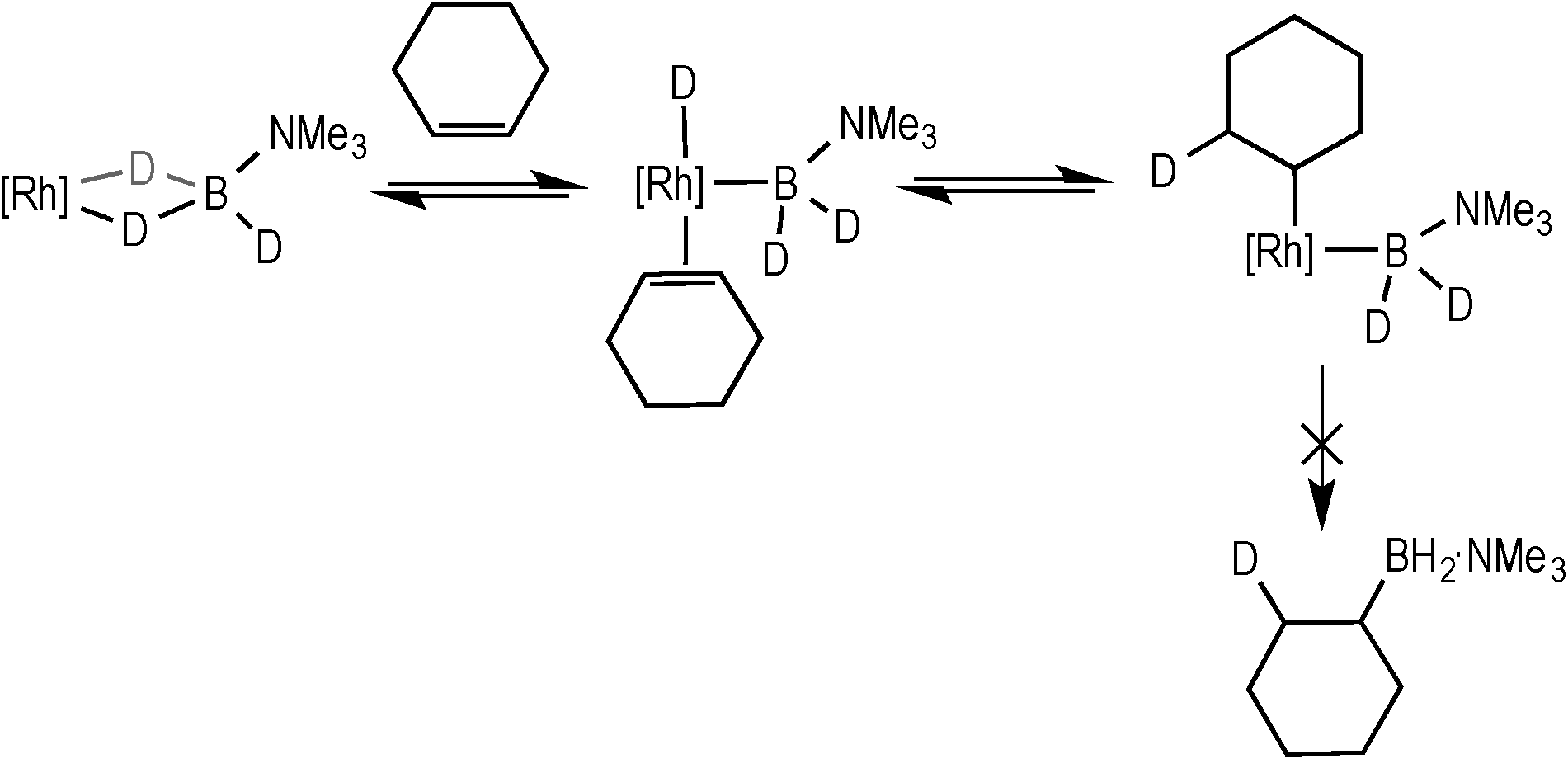 | ||
| Scheme 7 Reversible deuterio-insertion with cyclohexene. | ||
With 1-hexene, catalysis (5 mol% of 3 relative to H3B·NMe3) reached 37% conversion after 30 minutes yielding a product consistent with Me(CH2)5H2B·NMe3, as shown by a triplet [J(HB) = 91 Hz] in the 11B NMR spectrum at δ −1.4, although we were not able to isolate this material pure and thus cannot comment on the linear:branched ratio. After 1 week, a maximum conversion of 63% is reached. However, significant decomposition of the catalyst was observed, for which we cannot definitively provide a structure derived from the spectroscopic data. Thus, the hydroboration of alkenes with 3 appears to work best with TBE, with other alkenes only of limited utility.
Hydroboration with phosphine–boranes
The addition of H2 to [Rh(xantphos)(NBD)][BArF4] (NBD = norbornadiene) in the presence of the tertiary phosphine–borane H3B·PCy3 afforded [Rh(κ3P,O,P-xantphos)(H)2(η1-H3B·PCy3)][BArF4] (5) in quantitative yield by NMR spectroscopy (Scheme 8). Complex 5 was characterised in situ using NMR spectroscopy, and presents very similar data to the analogous complex 1.18 It is also related to the sigma phosphine–borane complex [Ru(xantphos)(H)(PPh2H)(H3B·PPh2H)][BArF4].40 The 1H NMR spectrum shows 3 hydride environments in a 3:1:1 ratio, consistent with the phosphine–borane bound in an η1 fashion that is undergoing rapid exchange between B–H–Rh and B–H groups, and two mutually cis hydrides: δ −1.42 [br, BH3, sharpens on 11B decoupling], δ −14.62 [br dtd, RhH] and δ −19.13 [dtd, RhH]. Two 31P environments are observed in the 31P{1H} NMR spectrum at δ 41.8 [d, J(RhP) = 114 Hz] and δ 20.1 (br, PCy3) in a 2:1 ratio respectively. Attempts to crystallise 5 were unsuccessful, resulting in decomposition.
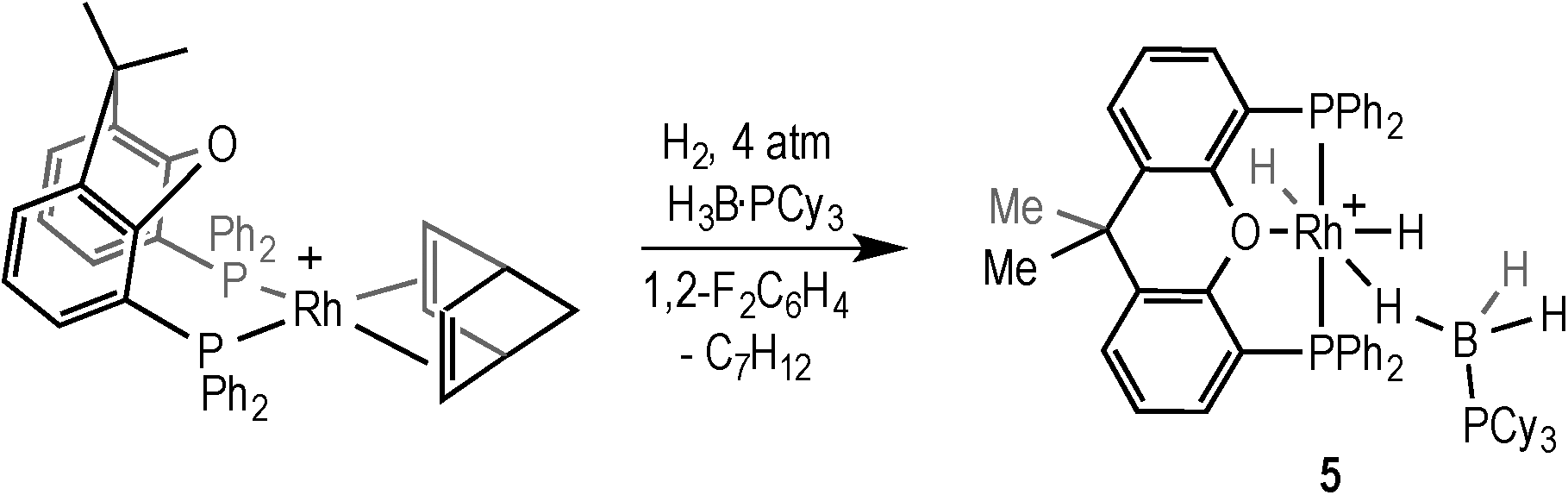 | ||
| Scheme 8 Formation of complex 5. [BArF4]− anion not shown. | ||
Addition of 2 equivalents of TBE to pale yellow 5 resulted in the formation of a dark green solution of a new compound formulated as [Rh(κ2P,P-xantphos)(η2-H3B·PCy3)][BArF4] (6), Scheme 9, in quantitative yield by NMR spectroscopy. Removal of volatiles allowed the isolation of 6 as a dark green solid. The NMR data for 6 are consistent with η2 binding of the phosphine–borane; in the 1H NMR spectrum, a quadrupolar broadened, integral 3H signal is observed at δ −2.38, which sharpens on 11B decoupling, while the 11B NMR spectrum shows a broad signal at δ −3.0. The 31P{1H} NMR spectrum is also consistent with a Rh(I) phosphine–borane complex, with two signals observed at δ 28.9 [d, J(RhP) = 190 Hz] and δ 17.3 (br, PCy3). These data are consistent with those reported for other [Rh(chelating phosphine)(H3B·PR3)]+ complexes.41 Addition of H2 (4 atm) to a CD2Cl2 solution of 6 reforms complex 5 in quantitative yields by NMR spectroscopy. Degassing a CD2Cl2 solution of 5 and placing under static vacuum for 4 hours resulted in an approximately 1:1 ratio of 5:6, suggestive of an equilibrium between the two species. Interestingly, for the [Rh(xantphos)]+ fragment we cannot isolate, or observe by NMR spectroscopy, the equivalent Rh(I) H3B·NMe3 complex to 6, as 4 forms instead from homocoupling.18
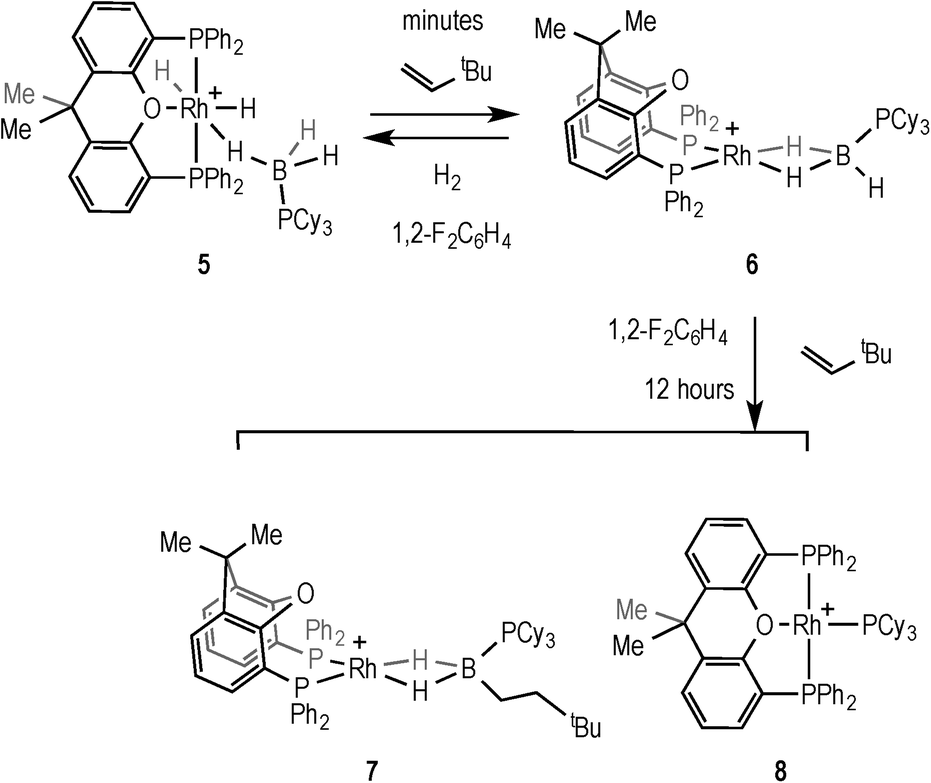 | ||
| Scheme 9 Formation of 6, 7 and 8. [BArF4]− anions are not shown. | ||
The ability of 6 to mediate hydroboration was probed by addition of excess (2.5 equiv.) TBE in 1,2-F2C6H4 solvent, by addition of the alkene to in situ generated 6, Scheme 9. After 45 minutes a new peak is apparent in the 1H NMR spectrum at δ −5.58 that is assigned to an Rh–HB interaction, consistent with the slow formation of [Rh(κ2P,P-xantphos)(η2-H2B(CH2CH2tBu)·PCy3)][BArF4] (7), similar to 2 and 3. After 16 hours the ratio of 7 had increased relative to 6 (~5:1 7:6). However small amounts (ca. 5% by 31P{1H} NMR spectroscopy) of a parallel product resulting from P–B cleavage [Rh(κ3P,O,P-xantphos)(PCy3)][BArF4] (8) were also observed by 31P{1H} NMR spectroscopy at δ 61.3 [dt, J(RhP) = 192, J(PP) = 34 Hz] and δ 37.8 [dd, J(RhP) = 155, J(PP) = 34 Hz]. P–B bond cleavage has been noted previously during metal-catalysed dehydrocoupling of phosphine–boranes.40,42,43 After a further 12 hours all of 6 was consumed, but a greater proportion of 8 (ca. 33%) was also present. Recrystallisation of the reaction mixture after several hours afforded a small crop of green crystals of 7 suitable for X-ray diffraction, and although the resulting data quality was poor and only the gross connectivity can thus be discussed, the solid-state structure of 7 suggests anti-Markovnikov hydroboration, as with 2 and 3 (see ESI†). The bulk composition could not be reliably determined by NMR spectroscopy as the alkyl region of the 1H NMR spectrum is dominated by the cyclohexyl peaks from the mixture of 6 and 8.
From such mixtures, several orange crystals of 8 also grew, confirming its solid-state structure (Fig. 4) as a square planar Rh(I) complex [sum of angles around Rh = 361.5°] with xantphos coordinated in a mer manner, similar to the closely related [Rh(κ3P,O,P-xantphos)(PCyp3)][BArF4] (Cyp = cyclopentyl),44 and [Rh(κ3P,O,P-xantphos)(CO)][BF4].45 Complex 8 has been independently synthesised from addition of PCy3 to 2 (see Experimental).
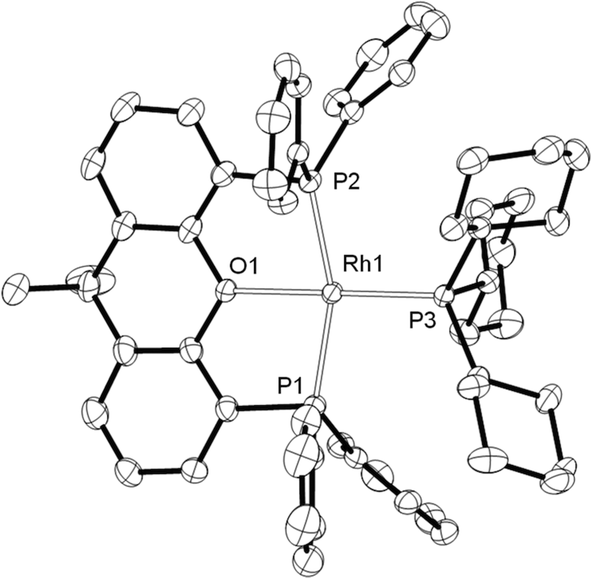 | ||
| Fig. 4 Solid state structure of the cationic portion of 8. [BArF4]− anion and H atoms are omitted for clarity. Displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): Rh1–P1,2.2854(7); Rh1–P2,2.2923(7); P3–Rh1, 2.2611(7); Rh1–O1, 2.2395(19); P2–Rh1–P1,159.70(3); P3–Rh1–O1, 170.44(6). | ||
Conclusions
We have described that the [Rh(xantphos)]+ fragment acts as an effective catalyst for the hydroboration of TBE using the amine–borane H3B·NMe3 at low (0.5 mol%) catalyst loadings. Investigations into the mechanism using the initial rate method and labelling studies show that reductive elimination of the linear hydroboration product is likely the rate-limiting step at the early stages of catalysis. This system is not as effective for other alkenes such as 1-hexene, or using the phosphine–borane H3B·PCy3; with decomposition or P–B bond cleavage occurring respectively.Experimental section
All manipulations, unless otherwise stated, were performed under an argon atmosphere using standard Schlenk and glove-box techniques. Glassware was oven dried at 130 °C overnight and flamed under vacuum prior to use. Pentane, hexanes, CH2Cl2 and MeCN were dried using a Grubbs type solvent purification system (MBraun SPS-800) and degassed by successive freeze–pump–thaw cycles. 1,2-F2C6H4 (pre-treated with alumina) and CD2Cl2 were dried over CaH2, vacuum distilled and stored over 3 Å molecular sieves. H3B·NMe3 was purchased from Aldrich and sublimed prior to use (5 × 10−2 Torr, 298 K). Cyclohexene, trimethylvinyl silane (TVMS), and 3,3-dimethyl-1-butene (TBE) were purchased from Aldrich, dried over sodium, vacuum distilled and stored over 3 Å molecular sieves. [Rh(xantphos)(NBD)][BArF4] was prepared by the literature method.46 NMR spectra were recorded on a Bruker AVIII-500 spectrometer at room temperature, unless otherwise stated. In 1,2-F2C6H4, 1H NMR spectra were pre-locked to a sample of C6D6 (25%) and 1,2-F2C6H4 (75%) and referenced to the centre of the downfield solvent multiplet, δ = 7.07. 31P and 11B NMR spectra were referenced against 85% H3PO4 (external) and BF3·OEt2 (external) respectively. Chemical shifts (δ) are quoted in ppm and coupling constants (J) in Hz. ESI-MS were recorded on a Bruker MicrOTOF instrument interfaced with a glove-box,28 or using the PSI-ESI technique as described previously,29,30 and in detail below. Microanalyses were performed by Elemental Microanalysis Ltd.ESI-MS reaction monitoring using pressurized sample infusion
A Schlenk flask under nitrogen containing 3 (4.7 mg, 0.0028 mmol) and H3B·NMe3 (1.4 mg, 0.019 mmol) was pressurized to 1.5 psi using 99.998% purity argon gas and connected to the mass spectrometer via a short length of PEEK tubing. A solution of TBE (4.8 μL, 0.038 mmol) in 1,2-F2C6H4 (3 mL) was injected into the pressurized Schlenk flask through a septum and collection on the mass spectrometer was initiated. Mass spectra were collected on a Micromass Q-Tof micro mass spectrometer in positive ion mode using pneumatically assisted electrospray ionization: capillary voltage, 2900 V; sample cone voltage, 15 V; extraction voltage, 0.5 V; source temperature, 92 °C; desolvation temperature, 192 °C; cone gas flow, 100 L h−1; desolvation gas flow, 200 L h−1; collision voltage, 2 V; MCP voltage, 2400 V. No smoothing of the data was performed. Aliquots of the reaction mixture were removed via syringe during the reaction for analysis by 11B NMR spectroscopy.General procedure for catalytic hydroboration
The alkene and 1,2-F2C6H4 (0.6 mL) were mixed in a Young's NMR tube and transferred to a new NMR tube containing 2 or 3 and H3B·NMe3. The samples were immediately frozen in liquid N2, and monitored in situ by 11B NMR spectroscopy on warming. See Table 1 for more details of relative concentrations.Synthesis and characterisation of new complexes
Acknowledgements
NSERC (Discovery and Discovery Accelerator Supplements) for operational funding and CFI and BCKDF for infrastructure (JSM); The University of Victoria for graduate scholarships (RT); the Royal Society for an International Exchange Award (ASW and JSM), the EPSRC for a DTG award (HCJ).Notes and references
- D. Mannig and H. Noth, Angew. Chem., Int. Ed. Engl., 1985, 24, 878–879 CrossRef.
- J. Hartwig, Organotransition metal chemistry: from bonding to catalysis, University Science Books, Sausalito, 2010 Search PubMed.
- C. M. Crudden and D. R. Edwards, Eur. J. Org. Chem., 2003, 2003, 4695–4712 CrossRef.
- T. Davan, E. W. Corcoran and L. G. Sneddon, Organometallics, 1983, 2, 1693–1694 CrossRef CAS.
- K. Burgess, W. A. Van der donk, S. A. Westcott, T. B. Marder, R. T. Baker and J. C. Calabrese, J. Am. Chem. Soc., 1992, 114, 9350–9359 CrossRef CAS.
- D. A. Evans, G. C. Fu and B. A. Anderson, J. Am. Chem. Soc., 1992, 114, 6679–6685 CrossRef CAS.
- D. R. Edwards, Y. B. Hleba, C. J. Lata, L. A. Calhoun and C. M. Crudden, Angew. Chem., Int. Ed., 2007, 46, 7799–7802 CrossRef CAS PubMed.
- J. M. Brown and G. C. Lloyd Jones, J. Am. Chem. Soc., 1994, 116, 866–878 CrossRef CAS.
- A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023–4078 CrossRef CAS PubMed.
- M. Scheideman, G. Wang and E. Vedejs, J. Am. Chem. Soc., 2008, 130, 8669–8676 CrossRef CAS PubMed.
- E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817–829 CrossRef CAS PubMed.
- L. J. Sewell, M. A. Huertos, M. E. Dickinson, A. S. Weller and G. C. Lloyd-Jones, Inorg. Chem., 2013, 52, 4509–4516 CrossRef CAS PubMed.
- L. J. Sewell, G. C. Lloyd-Jones and A. S. Weller, J. Am. Chem. Soc., 2012, 134, 3598–3610 CrossRef CAS PubMed.
- L. J. Sewell, A. B. Chaplin and A. S. Weller, Dalton Trans., 2011, 40, 7499–7501 RSC.
- In this report we did not describe H/D labelling studies. Subsequent studies using D3B·NMe3 showed that alkene coordination and deuteride insertion to form the linear product where all reversible, as found in this current study and consistent with reductive elimination being the rate-determining step at the early stages of catalysis. L. J. Sewell, DPhil Thesis, University of Oxford, 2013 Search PubMed.
- M. Toure, O. Chuzel and J.-L. Parrain, J. Am. Chem. Soc., 2012, 134, 17892–17895 CrossRef CAS PubMed.
- M. Kranenburg, Y. E. M. van der Burgt, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz and J. Fraanje, Organometallics, 1995, 14, 3081–3089 CrossRef CAS.
- H. C. Johnson, C. L. McMullin, S. D. Pike, S. A. Macgregor and A. S. Weller, Angew. Chem., Int. Ed., 2013, 52, 9776–9780 CrossRef CAS PubMed.
- T. M. Douglas, A. B. Chaplin, A. S. Weller, X. Yang and M. B. Hall, J. Am. Chem. Soc., 2009, 131, 15440–15456 CrossRef CAS PubMed.
- G. Alcaraz and S. Sabo-Etienne, Coord. Chem. Rev., 2008, 252, 2395–2409 CrossRef CAS PubMed.
- A. B. Chaplin and A. S. Weller, Inorg. Chem., 2010, 49, 1111–1121 CrossRef CAS PubMed.
- W. H. Bernskoetter, C. K. Schauer, K. I. Goldberg and M. Brookhart, Science, 2009, 326, 553–556 CrossRef CAS PubMed.
- M. D. Walter, P. S. White, C. K. Schauer and M. Brookhart, J. Am. Chem. Soc., 2013, 135, 15933–15947 CrossRef CAS PubMed.
- S. D. Pike, A. L. Thompson, A. G. Algarra, D. C. Apperley, S. A. Macgregor and A. S. Weller, Science, 2012, 337, 1648–1651 CrossRef CAS PubMed.
- Y. Kawano, K. Yamaguchi, S. Y. Miyake, T. Kakizawa and M. Shimoi, Chem. – Eur. J., 2007, 13, 6920–6931 CrossRef CAS PubMed.
- Related Rh(PR3)2(η2-carborane) complexes have been reported as the resting states during cage B-H hydroboration reactions with alkenes: J. D. Hewes, C. W. Kreimendahl, T. B. Marder and M. F. Hawthorne, J. Am. Chem. Soc., 1984, 106, 5757–5759 CrossRef CAS; E. Molinos, S. K. Brayshaw, G. Kociok-Köhn and A. S. Weller, Organometallics, 2007, 26, 2370–2382 CrossRef.
- R. B. Jordan, Reaction Mechanisms of Inorganic and Organometallic Systems, Oxford University Press, 2007 Search PubMed.
- A. T. Lubben, J. S. McIndoe and A. S. Weller, Organometallics, 2008, 27, 3303–3306 CrossRef CAS.
- K. L. Vikse, M. P. Woods and J. S. McIndoe, Organometallics, 2010, 29, 6615–6618 CrossRef CAS.
- L. P. E. Yunker, R. L. Stoddard and J. S. McIndoe, J. Mass Spectrom., 2014, 49, 1–8 CrossRef CAS PubMed.
- A. B. Chaplin and A. S. Weller, Angew. Chem., Int. Ed., 2010, 49, 581–584 CrossRef CAS PubMed.
- A. G. Algarra, L. J. Sewell, H. C. Johnson, S. A. Macgregor and A. S. Weller, Dalton Trans., 2014 10.1039/c3dt52771a.
- Reliable integration to quantify the degree of H/D exchange in the borane was frustrated by the quadrupolar-broadened signals.
- R. T. Baker, J. C. Calabrese, S. A. Westcott, P. Nguyen and T. B. Marder, J. Am. Chem. Soc., 1993, 115, 4367–4368 CrossRef CAS.
- D. E. Kadlecek, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 2000, 122, 10868–10877 CrossRef CAS.
- E. Molinos, S. K. Brayshaw, G. Kociok-Köhn and A. S. Weller, Organometallics, 2007, 26, 2370–2382 CrossRef CAS.
- T. C. Morrill, C. A. D'Souza, L. Yang and A. J. Sampognaro, J. Org. Chem., 2002, 67, 2481–2484 CrossRef CAS PubMed.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- R. Ghosh, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2008, 130, 11317–11327 CrossRef CAS PubMed.
- A. E. W. Ledger, C. E. Ellul, M. F. Mahon, J. M. J. Williams and M. K. Whittlesey, Chem. – Eur. J., 2011, 17, 8704–8713 CrossRef CAS PubMed.
- M. A. Huertos and A. S. Weller, Chem. Sci., 2013, 4, 1881 RSC.
- M. A. Huertos and A. S. Weller, Chem. Commun., 2012, 48, 7185–7187 RSC.
- T. N. Hooper, M. A. Huertos, T. Jurca, S. D. Pike, A. S. Weller and I. Manners, Inorg. Chem., 2014, 53, 3716–3729 CrossRef CAS PubMed.
- R. Dallanegra, A. B. Chaplin and A. S. Weller, Organometallics, 2012, 31, 2720–2728 CrossRef CAS.
- A. J. Sandee, L. A. van der Veen, J. Reek, P. Kamer, M. Lutz, A. L. Spek and P. van Leeuwen, Angew. Chem., Int. Ed., 1999, 38, 3231–3235 CrossRef CAS.
- R. J. Pawley, G. L. Moxham, R. Dallanegra, A. B. Chaplin, S. K. Brayshaw, A. S. Weller and M. C. Willis, Organometallics, 2010, 29, 1717–1728 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data collection and refinement details. Crystallographic data have been deposited with the Cambridge Crystallographic Data Center (CCDC), CCDC 1001382 (3) and 1001383 (8). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cy00597j |
| This journal is © The Royal Society of Chemistry 2014 |