 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The kinetics and product state distributions from gas-phase reactions of small atomic and molecular cations with C2H4, C2H3F, 1,1-C2H2F2, C2HF3 and C2F4†
Michael A.
Parkes‡
a,
Matthew J.
Simpson§
a,
Victor
Mikhailov¶
b and
Richard P.
Tuckett
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: r.p.tuckett@bham.ac.uk; Fax: +44 (0)121 414 4403; Tel: +44 (0)121 414 4425
bSchool of Physics and Astronomy, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK
First published on 7th January 2014
Abstract
The reactions of twenty one gas-phase cations with C2H3F, 1,1-C2H2F2, C2HF3 and C2F4 have been studied in a selected ion flow tube at 298 K. The cations are both atomic and molecular with recombination energies in the range 6–22 eV, and the kinetics and branching ratios into product ions are revealed for all the reactions. These data, together with that from an earlier study of reactions of CxFy+ with these four fluorinated ethenes (J. Phys. Chem. A., 2012, 116, 8119), are compared with the reactions of these ions with C2H4, where available. Nearly all the reactions have a rate coefficient close to the collisional value calculated by either Langevin or modified average dipole orientation theories. The products of the reactions of N+ and N2+ with C2H4 are found to be anomalous, compared to their reactions with the four fluorinated ethenes. The branching ratios into product cations are compared with those from a high resolution (ca. 0.002 eV) photoionisation (hν = 10–22 eV) study of C2H3F, 1,1-C2H2F2, C2HF3 and C2F4 (Phys. Chem. Chem. Phys., 2012, 14, 3935) in order to gauge the importance of electron transfer in ion–molecule reactions. The higher the recombination energy of the cation, the better the agreement between the two sets of product branching ratios. Where there is disagreement at lower recombination energies, it appears that there is more fragmentation of the products in the photoionisation experiment compared to the ion–molecule reactions.
1. Introduction
Following the acceptance of the 1987 Montreal Protocol,1 hydrofluorocarbons (HFCs) have been used as substitutes for chlorofluorocarbons in applications such as refrigeration, aerosols and fire retardants. Having no chlorine atoms, HFCs are more environmentally friendly to the stratosphere where ozone depletion occurs. However, they still have the capacity to contribute significantly to global warming in the boundary region and the troposphere due to their strong IR absorption in the range 8–13 μm, relatively long lifetimes and hence high global warming potential values.2 HFCs are removed from the earth's atmosphere predominantly by reaction with the OH radical in the troposphere. This study investigates the reactions of a series of fluorinated ethenes, C2HxF4−x (x = 4, 3, 2, 1, 0), with a range of small atomic and molecular cations in the gas phase. The rate coefficients and product state distributions of product cations are measured in a selected ion flow tube (SIFT) at 298 K. Whilst not of immediate relevance to the environmental aspects of HFCs presence in the earth's atmosphere, much of the data presented for ethenes with one, two or three fluorine atoms is new. Data for reactions of C2H4 and C2F4 come from elsewhere,3–21 and much of it is collected in the review of Anicich.22 This work extends similar studies on the chloroethene series including the three isomers of dichloroethene,23,24 and of the reactions of the fluorinated ethenes with CF+, CF2+, CF3+ and C2F4+.25 The reagent ions in this study range from H3O+, with the lowest recombination energy (RE) of 6.27 eV, through to Ne+ with the highest of 21.56 eV. (The RE is the energy released when a cation gains an electron to form the neutral atom or molecule, so its value is taken as the adiabatic ionisation energy (IE) of the neutral species.) The 1,2-isomers of C2H2F2 are less stable than the 1,1-isomer by ca. 50 kJ mol−1,26 and are not readily available. Hence the reactions of these two isomers have not been studied in this work. Using ion cyclotron resonance mass spectrometry (ICRMS), Bowers et al. have also studied a limited number of the reactions of the fluorinated ethenes with small molecular cations.27–31 Where available, our results are compared with these previous studies.This study compares a series of ion–molecule (IM) reactions from a fundamental perspective, the main aim being to see what effect the systematic replacement of hydrogen with fluorine atoms in ethene has on both the kinetics and the product branching ratios (BRs). Whilst the rate coefficients for IM reactions can be predicted theoretically (Section 3.1), the BRs are in general beyond theoretical calculation. Therefore, a comparison of BRs with another experimental method, photoionisation, is the only means currently available to learn more about the dynamics of the IM reactions. The adiabatic IE values of C2H4, C2H3F, 1,1-C2H2F2, C2HF3 and C2F4 are 10.51, 10.36, 10.30, 10.14 and 10.11 eV, respectively,32,33 the decrease of IE with increasing fluorination showing one aspect of the perfluoro effect.34 A comparison of these values with the RE of the reactant ion determines if charge transfer is energetically possible. Thus charge transfer is possible if RE (ion) exceeds ca. 10.5 eV, but is not possible for H3O+, SF3+ (RE = 8.32 eV), NO+ (9.26 eV), SF5+ (9.78 eV), SF2+ (10.24 eV) and SF+ (10.31 eV). If charge transfer is allowed and if the electron jump occurs at long distance when the intermolecular interaction between the reactant ion and the HFC neutral molecule is small, then one might expect the HFC cation to be formed with the same amount of electronic and vibrational energy as if it was formed by photoionisation with a photon whose energy is equal to the RE (ion),
i.e.
| AB+ (RE = y) + C2HxF4−x → (C2HxF4−x+)* + AB |
| (C2HxF4−x+)* → fragment ions + neutrals | (1) |
and
| C2HxF4−x + hν (energy = y) → (C2HxF4−x+)* + e− |
| (C2HxF4−x+)* → fragment ions + neutrals | (2) |
It is noted, however, that this argument makes the assumption that the neutralised molecular ion, AB, is not formed electronically, vibrationally or rotationally excited to any significant extent. In this scenario, the method of production of (C2HxF4−x+)* is immaterial, and one would expect similar product state distributions of fragment cations from the IM and the photoionisation experiments.3 Furthermore, Franck–Condon (FC) factors for ionisation of the fluoroethene should be important, and long-range charge transfer is unlikely to be efficient if the RE of the reactant ion corresponds to a FC gap, i.e. where the overlap of vibrational wavefunctions is highly unfavourable and FC factors are very small. However, if the electron jump occurs at a shorter separation where there is some degree of interaction between the potential energy surfaces of reactant cation and fluoroethene, this may cause modification to the vibrational wavefunctions and associated FC factors; charge transfer can then be efficient at the energy of a FC gap, and there may be significant differences in the product state distributions from the IM and the photoionisation experiments. The former distributions are measured in the SIFT apparatus, the latter by (threshold) photoelectron photoion coincidence ((T)PEPICO) spectroscopy. We have recently made high resolution studies of such processes by TPEPICO spectroscopy for C2HxF4−x (x = 3, 2, 1, 0) on the vacuum-ultraviolet beamline of the third generation synchrotron at the Swiss Light Source.35 In this paper, we compare the product state distributions of fragment ions from the two experiments to determine the possible importance or otherwise of long-range charge transfer as the dominant mechanism of the IM reaction. Similar comparisons have been made before, including the chlorinated ethene series.23,24,36,37 These previous studies showed that the two sets of product state distributions were generally similar, although reactions of N+ (RE = 14.53 eV) were anomalous and always produced very different distributions.
2. Experimental
The reactions of twenty one atomic and molecular cations (in order of decreasing RE; Ne+, F+, Ar+, N2+, N+, CO+, Kr+, CO2+, O+, OH+, N2O+, H2O+, Xe+, O2+, SF4+, SF+, SF2+, SF5+, NO+, SF3+ and H3O+) with C2HxF4−x (x = 3, 2, 1) have been investigated at 298 K using a SIFT to determine rate coefficients, product ions and their BRs, and whether the ion is produced by a primary or a secondary reaction. Results are shown in Table 1. Results for the reactions of these fluorinated ethenes with four reactant fluorocarbon cations (CF+, CF2+, CF3+ and C2F4+) have been published elsewhere,25 but are shown in Table 1 for completeness. The data for the reaction of these twenty five cations with C2H4 and C2F4 have been taken from a range of papers in the literature,3–21 some of which describe earlier results taken with the Birmingham SIFT apparatus.| Cation | C2H4 (IE = 10.51 eV) α′ = 4.26, μD = 0, Kc = 1 | C2H3F (10.36) α′ = 3.99, μD = 1.47, Kc = 1.84 | 1,1-CH2CF2 (10.30) α′ = 5.01, μD = 1.39, Kc = 1.65 | C2HF3 (10.14) α′ = 4.16, μD = 1.32, Kc = 1.70 | C2F4 (10.11) α′ = 4.35, μD = 0, Kc = 1 |
|---|---|---|---|---|---|
| a m/z 65 detected could be a primary product CH2SF+, or a secondary product C2H3F2+. If m/z 65 is the secondary product then the branching ratio for m/z 64 (C2H2F2+ or CHSF+) will be 100%. b Isomeric forms of these two product species are not know, however it is proposed that both cation and neutral have the lowest-energy, 1,1-isomeric form. c Only the 1,1-isomer of C2H2F2 as product gives an exothermic reaction; both isomers of 1,2-C2H2F2 as products are endothermic reactions. d No reaction means kexp < ca. 10−13 cm3 molecule−1 s−1. e Where adducts are formed, the rate coefficient for the reaction A+ + B + M → AB+ + M will be pressure dependent. All our measurements are made at total pressures of ca. 0.5–1.0 mbar, well below the high-pressure limit. The second order rate coefficient quoted is the (pressure-independent) third order rate coefficient multiplied by the total pressure. | |||||
| Ne+ (RE = 21.56 eV) |
k
exp = 1.2 × 10−9
k c = 1.4 × 10−9 |
k
exp = 2.2 × 10−9
k c = 2.3 × 10−9 |
k
exp = 2.0 × 10−9
k c = 2.2 × 10−9 |
k
exp = 2.0 × 10−9
k c = 2.0 × 10−9 |
k
exp = 1.2 × 10−9
k c = 1.2 × 10−9 |
|
C2H3+ (82%) + H + Ne [−799]
C2H4+ (18%) + Ne [−1066] |
C2H2+ (60%)
C2HF+ (13%) C2H3+ (11%) + F + Ne [−745] CF+ (4%) CHF+ (4%) + CH2 + Ne [−449] C2H2F+ (3%) + H + Ne [−749] C2H3F+ (2%) + Ne [−1080] C2H+ (2%) CH2F+ (1%) |
CF+ (34%)
C2HF+ (25%) C2H2+ (18%) CH2+ (13%) C2H2F+ (8%) + F + Ne [−677] CF2+ (2%) + CH2 + Ne [−446] |
CF+ (60%)
C2HF2+ (13%) + F + Ne [−657] CF2+ (11%) + CHF + Ne [−541] C2HF+ (7%) CHF+ (7%) CHF2+ (1%) C2F+ (1%) |
CF+ (80%)
CF2+ (15%) C2F4+ (4%) + Ne [−1105] CF3+ (1%) |
|
| Ref. 20 | Ref. 3 | ||||
| F+ (17.42) |
k
exp = 1.4 × 10−9
k c = 1.4 × 10−9 |
Reaction not studied |
k
exp = 2.4 × 10−9
k c = 2.3 × 10−9 |
k
exp = 2.5 × 10−9
k c = 2.1 × 10−9 |
k
exp = 1.1 × 10−9
k c = 1.2 × 10−9 |
|
C2H3+ (66%) + H + F [−400]
C2H2+ (28%) + H2 + F [−406] C2H4+ (6%) + F [−667] |
C2H2F+ (45%) + F + F [−278]
CH2F+ (28%) + CF + F [−246] CF+ (18%) + CH2F + F [−239] C2HF2+ (5%) + H + F [−267] C2H2F2+ (4%) + F [−687] |
Products not identified |
CF3+ (40%) + CF + F [−354]
CF2+ (38%) + CF2 + F [−310] C2F4+ (22%) + F [−705] |
||
| Ref. 12 and 13 | Ref. 3 | ||||
| Ar+ (15.76) |
k
exp = 1.1 × 10−9
k c = 1.2 × 10−9 |
k
exp = 1.8 × 10−9
k c = 1.9 × 10−9 |
k
exp = 1.8 × 10−9
k c = 1.8 × 10−9 |
k
exp = 1.6 × 10−9
k c = 1.6 × 10−9 |
k
exp = 0.88 × 10−9
k c = 0.91 × 10−9 |
|
C2H3+ (76%) + H + Ar [−240]
C2H2+ (20%) + H2 + Ar [−247] C2H4+ (4%) + Ar [−507] |
C2H3+ (57%) + F + Ar [−186]
C2H2F+ (18%) + H + Ar [−190] C2H2+ (12%) + HF + Ar [−326] C2HF+ (7%) + H2 + Ar [−188] CF+ (5%) + CH3 + Ar [−113] C2H3F+ (1%) + Ar [−521] |
C2H2F+ (44%) + F + Ar [−118]
CH2F+ (22%) + CF + Ar [−87] C2HF+ (19%) + HF + Ar [−250] CF+ (12%) + CH2F + Ar [−80] C2H2F2+ (3%) + Ar [−527] |
CHF2+ (51%) + CF + Ar [−180]
CHF+ (20%) + CF2 + Ar [−117] C2HF2+ (13%) + F + Ar [−98] CF+ (8%) + CHF2 + Ar [−149] C2HF3+ (4%) + Ar [−543] C2F2+ (4%) + HF + Ar [−194] |
CF2+ (28%) + CF2 + Ar [−150]
CF3+ (27%) + CF + Ar [−194] C2F3+ (24%) + F + Ar [+15] CF+ (17%) + CF3 + Ar [−192] C2F4+ (4%) + Ar [−546] |
|
| Ref. 10 | Ref. 3 | ||||
| N2+ (15.58) |
k
exp = 1.3 × 10−9
k c = 1.3 × 10−9 |
k
exp = 1.7 × 10−9
k c = 2.1 × 10−9 |
k
exp = 2.0 × 10−9
k c = 2.0 × 10−9 |
k
exp = 1.8 × 10−9
k c = 1.8 × 10−9 |
k
exp = 1.1 × 10−9
k c = 1.0 × 10−9 |
|
C2H3+ (50%) + H + N2 [−222]
C2H2+ (20%) + H2 + N2 [−229] HNC+ (10%) + HCN + H HCNH+ (10%) + HCN + H N2H+ (10%) + C2H3 |
C2H3+ (40%) + F + N2 [−168]
C2H2F+ (29%) + H + N2 [−172] C2H2+ (15%) + HF + N2 [−308] C2HF+ (12%) + H2 + N2 [−170] C2H3F+ (4%) + N2 [−503] |
C2H2F+ (33%) + F + N2 [−100]
CH2F+ (28%) + CF + N2 [−69] C2HF+ (20%) + HF + N2 [−232] CF+ (11%) + CH2F + N2 [−62] C2H2F2+ (8%) + N2 [−509] |
CHF2+ (66%) + CF + N2 [−162]
CF+ (16%) + CHF2 + N2 [−131] CHF+ (12%) + CF2 + N2 [−99] C2HF3+ (6%) + N2 [−525] |
C2F4+ (46%) + N2 [−528]
CF+ (25%) + CF3 + N2 [−174] CF3+ (13%) + CF + N2 [−176] CF2+ (11%) + CF2 + N2 [−132] C2F3+ (5%) + F + N2 [+33] |
|
| Ref. 4 and 5 | Ref. 8 | ||||
| N+ (14.53) |
k
exp = 1.3 × 10−9
k c = 1.6 × 10−9 |
k
exp = 1.8 × 10−9
k c = 2.6 × 10−9 |
k
exp = 2.4 × 10−9
k c = 2.6 × 10−9 |
k
exp = 2.3 × 10−9
k c = 2.3 × 10−9 |
k
exp = 1.6 × 10−9
k c = 1.4 × 10−9 |
|
C2H3+ (30%) + H + N [−121]
C2H4+ (25%) + N [−388] C2H2+ (10%) + H2 + N [−128] HCN+ + CH3 (15%) HCNH+ + CH2 (10%) CH2CN+ + H2 (10%) |
C2H3F+ (52%) + N [−402]
C2H2F+ (21%) + H + N [−71] C2H3+ (20%) + F + N [−67] C2HF+ (6%) + H2 + N [−69] C2H2+ (1%) + HF + N [−208] |
C2H2F2+ (82%) + N [−409]
C2H2F+ (16%) + F + N [−1] C2HF+ (2%) + HF + N [−132] |
C2HF3+ (100%) + N [−424] |
C2F4+ (85%) + N [−427]
CF+ (9%) + CF3 + N [−73] C2F3+ (4%) + FN [−169] (F + N endothermic by 134 kJ mol−1) CF2+ (1%) + CF2 + N [−32] CF3+ (1%) + CF + N [−240] |
|
| Ref. 17 and 18 | Ref. 8 | ||||
| CO+ (14.01) | No data in Anicich22 |
k
exp = 2.2 × 10−9
k c = 2.1 × 10−9 |
k
exp = 2.2 × 10−9
k c = 2.0 × 10−9 |
k
exp = 1.8 × 10−9
k c = 1.8 × 10−9 |
k
exp = 1.1 × 10−9
k c = 1.0 × 10−9 |
|
C2H3F+ (39%) + CO [−351]
C2H2+ (27%) + HF + CO [−46] C2H2F+ (16%) + H + CO [−20] C2H3+ (16%) + F + CO [−16] C2HF+ (2%) + H2 + CO [−18] |
C2H2F2+ (84%) + CO [−358]
C2H2F+ (16%) + FCO [−89] (F + CO endothermic by 51 kJ mol−1) |
CHF2+ (50%) + CF + CO [−10]
C2HF3+ (41%) + CO [−373] CF+ (9%) + CHF2 + CO [+20] |
C2F4+ (100%) + CO [−376] | ||
| Ref. 8 | |||||
| Kr+ (14.00) [and (Kr+)* (14.67)?] |
k
exp = 0.74 × 10−9
k c = 1.1 × 10−9 |
k
exp = 1.6 × 10−9
k c = 1.6 × 10−9 |
k
exp = 1.2 × 10−9
k c = 1.4 × 10−9 |
k
exp = 1.2 × 10−9
k c = 1.3 × 10−9 |
Reaction not studied |
|
C2H3+ (45%) + H + Kr [−70]
C2H2+ (45%) + H2 + Kr [−76] C2H4+ (10%) + Kr [−337] |
C2H3F+ (39%) + Kr [−350]
C2H2+ (25%) + HF + Kr [−156] C2H2F+ (23%) + H + Kr [−20] C2HF+ (7%) + H2 + Kr [−18] C2H3+ (6%) + F + Kr [−16] |
Products not identified |
C2HF3+ (86%) + Kr [−372]
CHF2+ (14%) + CF + Kr [−9] |
||
| Ref. 14 | |||||
| CO2+ (13.76) | No data in Anicich22 |
k
exp = 1.9 × 10−9
k c = 1.8 × 10−9 |
k
exp = 1.4 × 10−9
k c = 1.7 × 10−9 |
k
exp = 1.2 × 10−9
k c = 1.5 × 10−9 |
k
exp = 0.21 × 10−9 (0.14 × 10−9 at 390 K)
k c = 0.88 × 10−9 |
|
C2H3F+ (90%) + CO2 [−328]
C2H2+ (9%) + HF + CO2 [−134] C2H3+ (1%) + F + CO2 [+7] |
C2H2F2+ (100%) + CO2 [−335] | C2HF3+ (100%) + CO2 [−350] | C2F4+ (100%) + CO2 [−353] | ||
| Ref. 8 | |||||
| O+ (13.62) |
k
exp = 1.4 × 10−9
k c = 1.5 × 10−9 |
k
exp = 2.5 × 10−9
k c = 2.5 × 10−9 |
k
exp = 2.0 × 10−9
k c = 2.4 × 10−9 |
k
exp not measured
k c = 2.2 × 10−9 |
k
exp = 1.5 × 10−9
k c = 1.3 × 10−9 |
|
C2H2+ (80%) + H2 + O [−39]
C2H3+ (15%) + H + O [−33] C2H4+ (5%) + O [−300] |
C2H3F+ (100%) + O [−313] | Products not identified | C2HF3+ (100%) + O [−335] | C2F4+ (100%) + O [−338] | |
| Ref. 11 | Ref. 8 | ||||
| OH+ (13.25) | No data in Anicich22 |
k
exp = 2.4 × 10−9
k c = 2.5 × 10−9 |
Reaction not studied |
k
exp = 2.2 × 10−9
k c = 2.2 × 10−9 |
Reaction not studied |
| Products not identified | Products not identified | ||||
| N2O+ (12.89) | No data in Anicich22 |
k
exp = 1.5 × 10−9
k c = 1.8 × 10−9 |
k
exp = 1.4 × 10−9
k c = 1.7 × 10−9 |
k
exp = 1.1 × 10−9
k c = 1.5 × 10−9 |
k
exp = 0.33 × 10−9 (0.23 × 10−9 at 390 K)
k c = 0.88 × 10−9 |
| C2H3F+ (100%) + N2O [−243] | C2H2F2+ (100%) + N2O [−250] | C2HF3+ (100%) + N2O [−265] | C2F4+ (100%) + N2O [−268] | ||
| Ref. 8 | |||||
| H2O+ (12.62) |
k
exp = 1.5 × 10−9
k c = 1.5 × 10−9 |
k
exp = 2.4 × 10−9
k c = 2.4 × 10−9 |
Reaction not studied |
k
exp = 2.0 × 10−9
k c = 2.1 × 10−9 |
k
exp = 1.3 × 10−9
k c = 1.2 × 10−9 |
| C2H4+ (100%) + H2O [−203] | Products not identified | Products not identified | C2F4+ (100%) + H2O [−241] | ||
| Ref. 19 | Ref. 8 | ||||
| Xe+ (12.13) [and (Xe+)* (13.44)] |
k
exp = 0.85 × 10−9
k c = 1.0 × 10−9 |
k
exp = 1.4 × 10−9
k c = 1.5 × 10−9 |
k
exp = 8.0 × 10−10
k c = 1.3 × 10−9 |
k
exp = 8.0 × 10−10
k c = 1.1 × 10−9 |
Reaction not studied |
|
C2H4+ (75%) + Xe [−156]
C2H2+ (25%) + H2 + Xe [−22] |
C2H3F+ (100%) + Xe [−170] | C2H2F2+ (100%) + Xe [−177] | C2HF3+ (100%) + Xe [−192] | ||
| Ref. 14 | |||||
| O2+ (12.07) |
k
exp = 0.68 × 10−9
k c = 1.3 × 10−9 |
k
exp = 2.1 × 10−9
k c = 2.0 × 10−9 |
k
exp = 1.8 × 10−9
k c = 1.9 × 10−9 |
k
exp = 1.9 × 10−9
k c = 1.7 × 10−9 |
k
exp = 1.1 × 10−9
k c = 0.98 × 10−9 |
| C2H4+ (100%) + O2 [−151] | C2H3F+ (100%) + O2 [−164] | C2H2F2+ (100%) + O2 [−171] | C2HF3+ (100%) + O2 [−186] | C2F4+ (100%) + O2 [−189] | |
| Ref. 15 | Ref. 8 | ||||
| SF4+ (11.99) | No data in Anicich22 |
k
exp = 1.1 × 10−9
k c = 1.5 × 10−9 |
k
exp = 1.5 × 10−9
k c = 1.4 × 10−9 |
k
exp = 1.2 × 10−9
k c = 1.2 × 10−9 |
Reaction not studied |
| Products not identified | C2H2F2+ (100%) + SF4 [−164] (or CHSF+) | C2HF3+ (100%) + SF4 [−179] | |||
| CF2+ (11.36) |
k
exp = 1.1 × 10−9
k c = 1.1 × 10−9 |
k
exp = 1.8 × 10−9
k c = 1.8 × 10−9 |
k
exp = 1.6 × 10−9
k c = 1.6 × 10−9 |
k
exp = 1.5 × 10−9
k c = 1.5 × 10−9 |
k
exp = 1.1 × 10−9 (1.0 × 10−9 at 496 K)
k c = 0.85 × 10−9 |
|
C3H3F2+ (55%) + H [−?]
C2H4+ (45%) + CF2 [−82] |
C2H3F+ (88%) + CF2 [−96]
C2H3+ (12%) + CF3 [−111] |
C2H2F2+ (100%) + CF2 [−103] | C2HF3+ (100%) + CF2 [−118] | C2F4+ (100%) + CF2 [−121] | |
| Ref. 25 | Ref. 25 | Ref. 25 | Ref. 25 | Ref. 9 | |
| SF+ (10.31) | No data in Anicich22 |
k
exp = 1.6 × 10−9
k c = 1.8 × 10−9 |
k
exp = 1.4 × 10−9
k c = 1.6 × 10−9 |
k
exp = 1.4 × 10−9
k c = 1.4 × 10−9 |
Reaction not studied |
| Products not identified |
CH2SF+ (80%) + CF2a [−?] (or secondary C2H3F2+)
C2H2F2+ (20%) + SF [−2] (or secondary CHSF+) |
C2HF3+ (100%) + SF [−17] | |||
| SF2+ (10.24) | No data in Anicich22 | No reactiond | No reactiond | No reactiond | Reaction not studied |
| C2F4+ (10.11) |
k
exp = 0.7 × 10−9
k c = 1.0 × 10−9 |
k
exp = 0.6 × 10−9
k c = 1.5 × 10−9 |
k
exp = 0.7 × 10−9
k c = 1.4 × 10−9 |
k
exp = 0.2 × 10−9
k c = 1.2 × 10−9 |
k
exp = 0.02 × 10−9 (0.003 × 10−9 at 496 K)
k c = 0.69 × 10−9 |
|
C2H2F2+ (95%) + C2H2F2b [−62]
C3H3F2+ (5%) + CHF2 [−?] |
C2HF3+ (45%) + C2H2F2c [−34]
C3H3F2+ (40%) + CF3 [−?] C3H2F3+ (10%) + CHF2 [−?] C2H2F2+ (3%) + C2HF3 [−18] C4H3F5+ adduct (2%) [−?]e |
C4H2F6+ adduct (60%) [−?]e
C3H2F3+ (30%) + CF3 [−?] C3HF4+ (10%) + CHF2 [−?] |
C2HF3+ (72%) + C2F4 [+3]
C3HF4+ (28%) + CF3 [−?] |
C3F5+ (100%) + CF3 [−12] | |
| Ref. 25 | Ref. 25 | Ref. 25 | Ref. 25 | Ref. 9 and 21 | |
| SF5+ (9.78) | No data in Anicich22 |
k
exp = 6.4 × 10−10
k c = 1.5 × 10−9 |
k
exp = 1.0 × 10−10
k c = 1.3 × 10−9 |
No reactiond | Reaction not studied |
| Products not identified |
SF3+ (53%) + CH2F–CF3 [−223]
C2H2F3+ (32%) + SF4 [+59] (or CHSF2+ + ?) C2H2F2+ (15%) + SF5 [+50] (or CHSF+ + ?) |
||||
| NO+ (9.26) | No data in Anicich22 | No reactiond | No reactiond | No reactiond | Reaction not studied |
| CF+ (9.11) |
k
exp = 1.1 × 10−9
k c = 1.3 × 10−9 |
k
exp = 2.1 × 10−9
k c = 2.0 × 10−9 |
k
exp = 1.4 × 10−9
k c = 1.9 × 10−9 |
k
exp = 1.0 × 10−9
k c = 1.7 × 10−9 |
k
exp = 0.61 × 10−9 (0.23 × 10−9 at 496 K)
k c = 1.0 × 10−9 |
|
CH2F+ (80%) + C2H2 [−106]
C3H3+ (20%) + HF [−268] |
C2H3+ (88%) + CF2 [−69]
CHF2+ (12%) + C2H2 [−156] |
CF3+ (88%) + C2H2 [−134]
C2H2F+ (7%) + CF2 [+2] CHF2+ (5%) + HCCF [−66] |
CF3+ (100%) + HCCF [−106] |
CF3+ (65%) + FCCF [−19]
C3F5+ adduct (32%) [−?]e C2F4+ (4%) + CF [+97] |
|
| Ref. 25 | Ref. 25 | Ref. 25 | Ref. 25 | Ref. 9 | |
| CF3+ (9.09) |
k
exp = 0.7 × 10−9
k c = 1.1 × 10−9 |
k
exp = 1.3 × 10−9
k c = 1.6 × 10−9 |
k
exp = 0.7 × 10−9
k c = 1.5 × 10−9 |
k
exp = 0.2 × 10−9
k c = 1.3 × 10−9 |
k
exp = 0.03 × 10−9 (0.005 × 10−9 at 496 K)
k c = 0.76 × 10−9 |
|
C3H3F2+ (60%) + HF [−?]
C2H3+ (40%) + CHF3 [−42] |
C2H3+ (75%) + CF4 [−92]
CHF2+ (25%) + C2H2F2c [−22] |
C2H2F+ (50%) + CF4 [−21]
C3H2F5+ adduct (44%) [−?]e C3HF4+ (6%) + HF [−?] |
C3HF6+ adduct (100%) [−?]e |
C3F7+ adduct (94%) [−?]e
C3F5+ (4%) + F2 [+346] C2F3+ (2%) + CF4 [+113] |
|
| Ref. 25 | Ref. 25 | Ref. 25 | Ref. 25 | Ref. 9 | |
| SF3+ (8.32) | No data in Anicich22 | No reactiond | No reactiond | No reactiond | Reaction not studied |
| H3O+ (6.27) |
k
exp = 0.08 × 10−9
k c = 1.4 × 10−9 |
k
exp = 2.3 × 10−9
k c = 2.4 × 10−9 |
k
exp = 2.3 × 10−9
k c = 2.3 × 10−9 |
Reaction not studied | Reaction not studied |
|
C2H5+ (65%) + H2O [+17]
C2H7O+ adduct (35%) [−?]e |
[C2H3F⋯H]+ (100%) + H2O [−34] | [C2H2F2⋯H]+ (100%) + H2O [−25] | |||
| Ref. 6, 7 and 16 | |||||
The SIFT technique has been described in detail elsewhere.38 The reactant cations were generated from an appropriate precursor (e.g. Ne, C2F6, H2O, SF6etc.) in a high pressure, ca. 10−4 mbar, electron ionisation source. A quadrupole mass filter was used to select the reagent ion before injection into a flow tube, 1 m in length and 8 cm in diameter. The carrier gas was He at a pressure of ca. 0.5 Torr, flowing at a velocity of ca. 100 m s−1. Conditions inside the flow tube were thermalised at 298 K, and any excited ions produced in the source should be collisionally cooled by the buffer gas. At a known distance downstream in the flow tube the neutral reactant gas was injected. The reaction gas mixture was sampled at the end of the flow tube through a 1 mm orifice in a Faraday plate. Reactant and product ions were focused into a second quadrupole mass filter and detected by an off-axis channeltron electron multiplier.
The experimental rate coefficients, kexp, were measured under pseudo-first-order conditions by recording the loss of reagent ion as a function of the concentration of neutral reagent. The measurement of the latter's absolute concentration, described elsewhere,39 is non-trivial and crucial to the accuracy of the result. The uncertainty in kexp values is estimated conservatively to be ±15%, and the apparatus is limited to measuring reactions with kexp ≥ ca. 10−13 cm3 molecule−1 s−1. Product ion BRs were obtained by recording their signals as a function of concentration of the neutral reagent. The ion signals were then extrapolated to zero concentration to give the BRs, which also allows identification of any secondary ion products. The conservative errors in the BRs are considered to be ±20%, although this value will be greater for minor products when BRs are below 10%.
Samples of C2H3F (98%) and C2HF3 (97%) were purchased from Apollo Scientific, 1,1-CH2CF2 (99+%) from Aldrich, and C2F4 (99+%) from Fluorochem. All gases were used without further purification.
3. Results and discussion
3.1 Kinetics and product state distributions
Our results are presented in columns 3–5 of Table 1. Columns 2 and 6 of the table show data from other sources for the reactions of C2H4 and C2F4. In addition to the experimental rate coefficient, kexp, product cations and their BRs, neutral products associated with the product cations are proposed. The corresponding reaction enthalpies, ΔrHo298, were calculated using enthalpies of formation for reactant and product species taken from standard sources.26,40 For cations at this temperature of 298 K, the stationary electron (or ion) convention is used.26 Updated ΔfHo298 values for ethene and the four neutral fluorinated ethene molecules (C2H4 +52.5, C2H3F −140.1, 1,1-C2H2F2 −350.2, C2HF3 −499.1, C2F4 −672.8 kJ mol−1) were taken from the recent high resolution TPEPICO study.35Collisional rate coefficients, kc, are also shown in Table 1. The ratio of kexp to kc gives the efficiency of the reaction, with values measured in this study spanning ca. 0.03 to 1.00. The kc values were calculated using Langevin capture theory for ions reacting with the non-polar molecules C2H4 and C2F4,41,42 and by the modified average dipole orientation (MADO) model for the polar molecules C2H3F, 1,1-C2H2F2 and C2HF3.43 The former theory calculates the rate coefficient for an R−4 attractive potential between a positive cation and a polarisable but non-polar neutral molecule; the rate coefficient is found to be independent of temperature. The latter theory is a modification to Langevin theory for polar neutral molecules, giving rise to both a larger rate coefficient and a small T−0.5 negative temperature dependence of the coefficient. The parameterised form of the Langevin rate coefficient for ions reacting with non-polar molecules is given by kL = 2.342 × 10−9 (α′/μ)1/2 cm3 molecule−1 s−1, where α′ is the polarisability volume of the neutral molecule in units of Å3 and μ is the reduced mass of the reacting pair in u. When the neutral molecule is polar, kL is multiplied by a dimensionless parameter, Kc, to give the MADO collisional rate coefficient. The Kc value is mildly temperature dependent, is given in the top row of Table 1 for the five molecules under study, and depends upon another dimensionless parameter, x, given in parameterised form at 298 K by x = 3.487 μD/(α′)1/2, where μD is the dipole moment of the polar molecule in Debye (D) and α′, as above, is its polarisability volume in Å3. When x lies between 2.0 and 3.0, as here for C2H3F, 1,1-C2H2F2 and C2HF3, its value is related to Kc by Kc = 0.477x + 0.620;43 when x is zero, Kc takes the value of unity. We use α′ values for C2H4, C2H3F, 1,1-C2H2F2, C2HF3 and C2F4 of 4.25, 3.99, 5.01, 4.16 and 4.35 Å3, respectively.44,45 The dipole moments of C2H3F, 1,1-C2H2F2 and C2HF3 are 1.47, 1.39 and 1.32 D, respectively.44
There are huge amounts of data presented in Table 1, and we present only generalised summaries. First, of the 61 studied reactions the majority are observed to occur very close to the calculated collisional rate coefficient. This suggests that on kinetic grounds the dynamics of many of these reactions, especially the ions with high RE, are dominated by long-range charge transfer. There are some exceptions to this finding. In one case, F+ + C2HF3, the experimental rate coefficient is larger than the calculated value by 20%, but this is only just outside the estimated error in rate coefficients of ±15%. Perhaps more informative are those reactions where the ratio of kexp to kc is much less than unity, say ca. <0.5. For these cases the reaction could be slower than expected for one of several reasons. The RE of the cation could lead to long-range charge transfer not being favoured (e.g. the energy of the reaction falls into a FC gap) or the reaction is not energetically possible (e.g. when the RE of the reagent cation is less than the IE of the neutral). When RE > IE, in addition to long-range charge transfer, a short-range mechanism can also occur, and then the sterics of the reaction can become important. The importance of steric effects seems clear in some of the reactions of C2F4, especially the reactions with CO2+, N2O+, C2F4+ and CF3+ where a large number of heavy atoms are involved and kexp is significantly less than kc. It should also be noted that all these four reactions show a slight negative temperature dependence,8,9 suggesting that the Langevin model is failing on the grounds of both reaction efficiency and temperature dependency. When RE < IE, only chemical reactions involving the exchange of atoms and formation of new chemical bonds can occur. Clearly, the kinetics will then be dominated by dynamical issues (including steric effects) and energetics. Examples are the slow reactions C2F4+ + C2F4 and H3O+ + C2H4 which are both close to thermoneutral in forming the preferred products C3F5+ + CF3 and C2H5+ + H2O, respectively.6,9
Second, assuming that products can only form from an exothermic reaction and that entropic effects can be ignored, there are several examples of IM reactions where the identity of the accompanying neutral product(s) can unambiguously be deduced. Obviously, as the RE(ion) increases, more possibilities for the neutral products become available. For ions with RE values in the range ca. 13–16 eV, mainly because of the strength of the H–H and H–F bonds in H2 and HF, following charge transfer it is often only possible to form an observed product ion where two atoms are lost if these atoms combine to form a bond in the exit channel. For example, C2H2+ (12%) from Ar+ + C2H3F can only form with HF + Ar, and not with H + F + Ar. Likewise, HCCF+ (7%) from the same reaction can only form with H2 + Ar, and not with H + H + Ar. Sometimes a product ion can only form if the neutralised molecular reactant ion itself forms a new bond. For example, CO+ + 1,1-C2H2F2 forms CF![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2+ (16%), but energetically this reaction is only exothermic if the FCO neutral, rather than F + CO, is formed in addition. This observation suggests that there is at least some short-range component to the reactions between CO+ and C2H2F2, and would explain why the branching ratio for photoionisation of C2H2F2 at 14 eV is different (see Section 3.2).
CH2+ (16%), but energetically this reaction is only exothermic if the FCO neutral, rather than F + CO, is formed in addition. This observation suggests that there is at least some short-range component to the reactions between CO+ and C2H2F2, and would explain why the branching ratio for photoionisation of C2H2F2 at 14 eV is different (see Section 3.2).
Third, the reactions of N2+ and N+ with C2H4 are the only two studies where completely different products are observed in the bimolecular IM study compared to the photoionisation study. Thus N2+ (RE = 15.58 eV) + C2H4 produces C2H3+ (50%) and C2H2+ (20%) as the major products, presumably via dissociative charge transfer, but also HNC+, HCNH+ and N2H+ are detected with BRs totalling 30%.4,5 These latter species can only result from a chemical reaction occurring between the reactants in which new bonds break and form. Photoionisation of C2H4 at 15.58 eV only produces C2H3+ and C2H2+.46 Whilst the products HNC+ and HCNH+ share the same m/z values as C2H3+ and C2H4+ and it is not clear how these products are differentiated in the SIFT study of McEwan et al.,4 N2H+ with m/z 29 has a different mass to the other products. Therefore, this apparent anomalous behaviour of N2+ and N+, that charge transfer and chemical reaction with C2H4 are occurring simultaneously, is confirmed. In support, a study of the reaction N+ + C2H4 using the Birmingham SIFT apparatus also produced the anomalous ions HCN+, HCNH+ and CH2CN+ totalling 35% of the products, as well as the expected ions C2H4+, C2H3+ and C2H2+.17 In this paper Smith and Adams note that isotopic labelling of the reactants confirmed their assignment of the product ions to be correct, and the first two ions with m/z 27 and 28 are not due to additional sources of C2H3+ and C2H4+. This study of N+ + C2H4, independently confirmed by Rakshit,18 suggests strongly that the N2+ + C2H4 reaction does indeed produce product ions in which one or more nitrogen atoms are incorporated into the hydrocarbon via a chemical intermediate in which bonds form and break.
Fourth, the reactions of CO+ (RE = 14.01 eV) and Kr+ (RE = 14.00 eV) with near identical RE values are revealing. With C2H3F, the ion–molecule BRs are almost identical for these two ions, but they show significant differences from the photoionisation BRs (Section 3.2 and table in ESI†); for example, the parent ion is dominant (39%) in the IM reactions, but is absent in the photoionisation experiments. With C2HF3, however, although both cations produce C2HF3+ and CHF2+ the BRs are very different, and again they show very different BRs with photoionisation (Section 3.2). It is only for 1,1-C2H2F2 that comparable results between the two experiments are observed. It is noticeable that the mid-point energy of 14.005 eV does not fall in a FC gap for C2H3F, but does for C2HF3.35 Thus reactions of C2H3F with ions of RE of ca. 14.0 eV might appear to be more suitable for long-range charge transfer than reactions of C2HF3 (see Section 1). Yet, these results, despite near-identical RE values, suggest a different mechanism for the reactions of Kr+ and CO+, and that long-range charge transfer does not make an important contribution. Like reactions of Xe+,14 however, Kr+ may also exist in the SIFT apparatus in its excited spin–orbit state at 14.67 eV. This was not apparent in the study of Giles et al. because the same products were observed from Kr+ 2P3/2 and (Kr+)* 2P1/2 reacting with C2H4.14 If this is so, then the comparison of the product BRs from Kr+ from those with CO+ reacting with all the fluorinated ethenes becomes less significant.
Fifth, for ions with an RE below the IE of the fluoroethene with which they are reacting, products can only form via a chemical reaction in which old bonds break and new bonds form in one or more of the transition states that link reactants to products. Carbon-containing cations are particularly suitable for such reactions, and the fluorocarbon cations CF+, CF3+ and C2F4+, all with REs below 10.15 eV, fall into this category. F− abstraction is particularly favoured for the reactions of CF+ and CF3+ with C2H3F. This molecule has the highest dipole moment of the fluoroethenes, suggesting that these ions attack the electron-rich fluorine in C2H3F rather than the CC bond. In general, however, these cations react with the fluoroethene molecules via an SN2 mechanism to form a 3- or 4-carbon non-cyclic adduct that may subsequently fragment or re-arrange to form new products.25 CF2+ is an interesting reactant because, although it has an RE as high as 11.36 eV,47 it reacts with C2H4 and C2H3F via a chemical reaction and cleavage/formation of new bonds within a collision complex.35 The evidence for this comes from the difference in BRs from the IM and photoionisation experiments (table in ESI†). By contrast, from Table 1 and the ESI† there is good evidence that this ion reacts with 1,1-C2H2F2, C2HF3 and C2F4via long-range charge transfer as these three reactions produce only the parent ion C2H2F2+etc., exactly as formed by non-dissociative photoionisation with hν = 11.36 eV.35
Some of these fluorocarbon cation reactions were also studied twenty years ago by Morris et al. in a SIFT apparatus,9 but there are anomalies in the minor products that they report for the reaction of CF3+ and CF+ with C2F4. The major product of the former reaction is the adduct C3F7+ (≥94%) which must form by an exothermic reaction. However, the minor products of C3F5+ (≤4%) and C2F3+ (≤2%) can only form with neutrals in their lowest energetic form, i.e. F2 and CF4, via endothermic reactions: +346 and +113 kJ mol−1, respectively. Likewise, whilst the major product from CF+ + C2F4, CF3+ (65%), can only form with neutral C2F2 in a mildly exothermic reaction, −19 kJ mol−1, and production of the C3F5+ (32%) adduct must be exothermic, the third product ion C2F4+ (4%) can only form with CF in a reaction which is endothermic by +97 kJ mol−1. These highly endothermic values cannot possibly result from errors in the thermochemical data for the individual species, and we suggest that these products resulted from experimental impurities or the presence of electronically excited reactant ions in the flow tube used by Morris et al.
Sixth, the ion with the lowest RE studied, H3O+ (6.27 eV), can only react with the fluorinated ethenes via a chemical reaction involving H+ transfer. Reactions with C2H3F and 1,1-C2H2F2 are fast and close to the collisional rate values, and the major product is indeed dissociative proton transfer with formation of H2O as the accompanying neutral providing a route for a mildly exothermic reaction, −34 and −25 kJ mol−1 respectively. However, the reaction of H3O+ with C2H4, measured in a SIFT apparatus by Matthews et al.,6 is much slower. Given this very low rate coefficient, it is somewhat surprising that the major products are again formed by dissociative proton transfer, C2H5+ + H2O (65%), with the adduct cation C2H7O+ being formed with a BR of 0.35. Using the recent stationary-electron value for ΔfHo298(C2H5+) from imaging PEPICO spectroscopy of 902.8 ± 1.3 kJ mol−1,48 this reaction is calculated to be +17 kJ mol−1 endothermic, and this may be sufficient reason to explain the low reaction efficiency (kexp/kc) of only 0.06 of the former reaction. Using the earlier Lias et al. value for ΔfHo298(C2H5+) of 914 ± 4 kJ mol−1,26 the reaction would then be as much as 28 kJ mol−1 endothermic, the reaction would then have a negligible rate coefficient and this is confirmation that this earlier value is too positive. So long as the reaction is exothermic and with the exception of some delocalised carbon ring molecules,49 the huge majority of reactions involving proton transfer proceed at or close to the collisional rate.50,51 This statement is not true for electron (or charge) transfer reactions.3 Furthermore, unlike electron transfer where the charge can transfer between the two reacting species at relatively long range, proton transfer, on account of the larger mass, involves a closer, shorter range, and more intimate interaction between the ion (H3O+) and the neutral molecule in which the proton moves from one to the other.52
3.2 Comparison of the product state distributions with photoionisation branching ratios
The branching ratios for production of daughter ions following photoionisation of the four fluoroethenes have been measured from threshold to ca. 22 eV at the Swiss Light Source using a threshold photoelectron–photoion coincidence spectrometer.35,53 As the electrons are imaged on to a position-sensitive detector by velocity map imaging, a resolution as high as 0.002 eV can be achieved.33,35 Photoionisation of C2H4 from threshold up to 18 eV has previously been measured at lower resolution, ca. 0.06 eV,46 and the results have been compared with the breakdown diagram predicted by quasi-equilibrium theory.54 The data of Stockbauer and Inghram46 have already been used to discuss the results of dissociative charge transfer reactions at thermal energy of Ar+ with C2H4.10A comparison of the BRs from these photoionisation experiments at photon energies corresponding to the RE of a cation (e.g. 21.56 and 15.58 eV, corresponding to Ne+ and N2+, respectively) with those from the IM reactions (Section 3.1) are tabulated in the ESI.† Mass discrimination effects have not been allowed for in either experiment, but they are expected to be small. The breakdown diagrams for C2H3F, 1,1-C2H2F2, C2HF3 and C2F4 are shown in Fig. 1–4 respectively, so that a comparison of the table in the ESI† with these figures can aid in comparing the BRs from the two experiments. The theoretically-determined breakdown diagram of C2H4 between 12 and 18 eV, taken from Fig. 1 of Bombach et al.54 is shown in Fig. 5. Such comparisons are, of course, only relevant if the RE(ion) exceeds the IE of the fluoroethene. As explained in the Introduction, a good agreement between the two sets of BRs might indicate that the dynamics of the bimolecular IM reaction are dominated by initial long-range electron transfer from the neutral fluorinated ethene molecule to the atomic/molecular cation. However, other mechanisms can also give similar BRs, so caution needs to be exercised not to over-interpret the data. With this caveat in mind, as with the kinetics data of Section 3.1, the table of the ESI† shows a huge amount of information and we make generic comments only.
 | ||
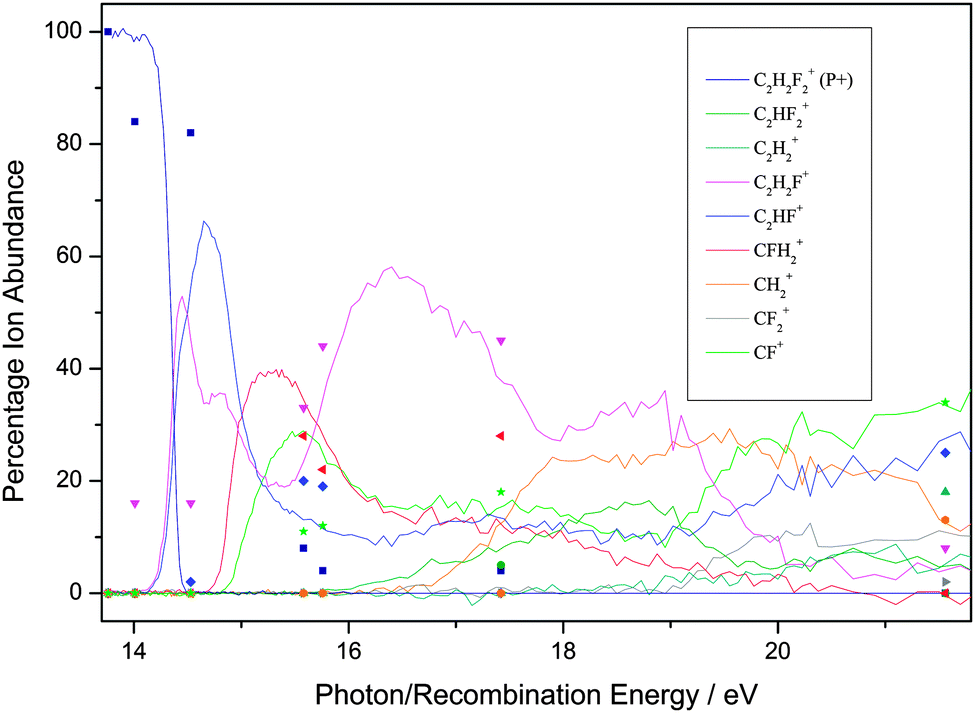
| Fig. 1 Breakdown diagram of C2H3F over the range 13–22 eV recorded with a (variable) resolution of 0.002 to 0.05 eV at the Swiss Light Source, Paul Scherrer Institute, Switzerland. The product branching ratios are accurate to ±1%. (Adapted from ref. 35, Phys. Chem. Chem. Phys., 2012, 14, 3935, with permission from the PCCP Owner Societies.) The diagram is compared with the branching ratios from the ion–molecule reactions at defined recombination energies of the reactant ion. | ||
 | ||
| Fig. 2 Breakdown diagram of 1,1-C2H2F2 over the range 14–22 eV recorded with a (variable) resolution of 0.002 to 0.05 eV at the Swiss Light Source, Paul Scherrer Institute, Switzerland. The product branching ratios are accurate to ±1%. (Adapted from ref. 35, Phys. Chem. Chem. Phys., 2012, 14, 3935, with permission from the PCCP Owner Societies.) The diagram is compared with the branching ratios from the ion–molecule reactions at defined recombination energies of the reactant ion. | ||
 | ||
| Fig. 3 Breakdown diagram of C2HF3 over the range 13–22 eV recorded with a (variable) resolution of 0.002 to 0.05 eV at the Swiss Light Source, Paul Scherrer Institute, Switzerland. The product branching ratios are accurate to ± 1%. (Adapted from ref. 35, Phys. Chem. Chem. Phys., 2012, 14, 3935, with permission from the PCCP Owner Societies.) The diagram is compared with the branching ratios from the ion–molecule reactions at defined recombination energies of the reactant ion. | ||
 | ||
| Fig. 4 Breakdown diagram of C2F4 over the range 13–19 eV recorded with a (variable) resolution of 0.002 to 0.05 eV at the Swiss Light Source, Paul Scherrer Institute, Switzerland. The product branching ratios are accurate to ± 2%. (Adapted from ref. 35, Phys. Chem. Chem. Phys., 2012, 14, 3935, with permission from the PCCP Owner Societies.) The diagram is compared with the branching ratios from the ion–molecule reactions at defined recombination energies of the reactant ion. | ||
 | ||
| Fig. 5 Calculated breakdown diagram (solid lines) of C2H4 over the range 12–18 eV recorded with a resolution of ca. 0.3 eV. Experimental values are taken from data of Stockbauer and Inghram,46J. Chem. Phys., 1975, 62, 4862. (Adapted, with permission, from Bombach et al.,54Int. J. Mass Spectrom. Ion Proc., 1984, 58, 217.) The diagram is compared with the branching ratios from the ion–molecule reactions at defined recombination energies of the reactant ion. | ||
First, for the rare gases Ne+, Ar+, Kr+ and Xe+, the RE of the ion exceeds the IE of the fluoroethene, so the process of charge transfer (CT) via an electron jump is energetically allowed. It is found that the agreement in BRs between the unimolecular photoionisation TPEPICO study and the bimolecular IM reaction tends to be good, but shows a tendency to degrade as the number of fluorine atoms in the fluoroethene increases. For the ions with the highest and lowest RE, Ne+ and Xe+, the agreement between BRs in the two experiments is the best. It is the worst for Kr+ with an intermediate RE of 14.00 eV.
Second, as might be expected, the ions with high RE values which easily exceed the IE of the fluoroethene tend to show the best agreement with the photoionisation BRs. There is a possible analogy with electron energy loss (e,e) dipole or ‘pseudo-photon’ spectroscopy: the higher the electron energy and hence the energy of the scattered electron, the more the technique resembles photon absorption spectroscopy, with hν being the difference in energy of the two electrons.55 However, with any one particular ion, e.g. F+, there seems to be no reason why some molecules give better agreement with the BRs than others. For example, the BRs for F+ + C2H4 show reasonable agreement whilst those for F+ + C2F4 show poor agreement, yet 17.42 eV falls in a FC gap in the photoelectron spectrum of both C2H4 and C2F4.35,56,58 With Ar+ (RE = 15.76 eV), however, the BR agreement is better with C2H4 where this energy is close to the maximum of the C2H4+ ![[C with combining tilde]](https://www.rsc.org/images/entities/char_0043_0303.gif) 2B3u state57 than with C2F4 where this energy is in the low-energy wing of the C2F4+ Ã 2Bg state.35,58 Yet with this same ion, the BR agreement is much better with C2H3F than with C2HF3, even though 15.76 eV corresponds in both molecules to an energy between ionic electronic states. In other words, whilst the presence of a FC gap is likely to reduce the rate of the reaction, it is not possible to determine a pattern whether the RE corresponds to a FC peak or a FC gap in the fluoroethene photoionisation spectrum is a significant factor in determining the IM branching ratios. This difficulty has been observed before.3
2B3u state57 than with C2F4 where this energy is in the low-energy wing of the C2F4+ Ã 2Bg state.35,58 Yet with this same ion, the BR agreement is much better with C2H3F than with C2HF3, even though 15.76 eV corresponds in both molecules to an energy between ionic electronic states. In other words, whilst the presence of a FC gap is likely to reduce the rate of the reaction, it is not possible to determine a pattern whether the RE corresponds to a FC peak or a FC gap in the fluoroethene photoionisation spectrum is a significant factor in determining the IM branching ratios. This difficulty has been observed before.3
Third, when disagreements between BRs from the two experiments are not within experimental error, the parent ion (P+) is always more intense in the IM reaction; for over ca. 20 reactions, the yield of P+(IM) significantly exceeds that of P+(hν), often by a factor as much as 5–10. The same phenomenon was observed with the related molecules C2H3Cl, C2HCl3 and C2Cl4,24 where data were recorded at the Daresbury Synchrotron Radiation Source at an inferior resolution, ca. 0.03 eV, compared to the Swiss Light Source. For the fluoroethene reactions there is only one exception; with CO+ + 1,1-C2H2F2, the BR(P+) is 84%, compared with 100% in the photon-induced reaction. Likewise, the larger fragment ions tend to have a higher BR from the IM reaction, whereas the photoionisation experiment tends to produce the smaller fragment ions with a higher BR. This suggests that for the IM reactions less energy is available to transfer to the fluoroethene than the absolute value of the RE(ion) would suggest. To some extent, this is not a surprising result as the neutralised molecular ion will almost certainly form over a range of vibrationally and rotationally excited states, and may even form in low-lying excited electronic states. If this occurs to a significant extent, then the BRs from the IM reactions and from photoionisation will appear to be shifted with respect to each other. Therefore, we conclude that taking the RE(ion) value as necessarily meaning the formation of ground state products may not be the most appropriate measure of the energy that is available for the reaction.
Fourth, as has been noted before in such comparisons of BRs from unimolecular photoionisation and bimolecular IM reactions, the reactions of N+ (RE = 14.53 eV) with these five ethene-like molecules produces very poor agreement. This fact has also been noted for molecules where the agreement with ions of RE < 14.5 eV and RE > 14.5 eV is generally good, but poor for N+,23,24,36,37 and it appears that N+ acts as a softly ionising species compared to photons of this energy. The argument above about vibrational and rotational excitation of the neutralised ion clearly cannot apply to an atomic ion. If, however, the product N atom is formed in an excited electronic state (e.g.2D lying 2.4 eV above the 4S ground state),59 then with this new lower and effective RE value, the agreement between the two experiments is much better. The reaction of N+ with C2H4 is clearly anomalous, as a chemical reaction occurs within a collision complex (see Section 3.1).
4. Conclusions
The kinetics and product state distributions of the reactions of fluorinated ethene molecules, ranging from zero to four fluorine atoms, with cations of RE in the range 6.27–21.56 eV have been studied. The branching ratios at twenty five specific values of the RE have been compared with those from photoionisation as measured by electron–ion coincidence spectroscopy. Difluoroethene can exist as three isomers; 1,1, cis-1,2 and trans-1,2. Unlike our earlier study of dichloroethene,23 however, it has not been possible to study if isotopic effects exist in the reactions of difluoroethene. Most of the results for reactions of mono-, di- and tri-fluoroethene with atomic and molecular cations are reported for the first time. The data for reactions of ethene and tetra-fluoroethene are collected together from other sources for comparison. Whilst some trends are apparent as the number of fluorine atoms in the substituted ethene molecule increases, especially for the reactions of CxFy+ where nucleophilic attack on the CC bond of the substituted ethene is the first step in the mechanism,25 it has proved difficult to formulate an overarching prescription for the dynamics/mechanism of these reactions. However, some general conclusions can be drawn.
The majority of the reactions occur at or close to the collisional value predicted by Langevin (for non-polar molecules) or MADO (for polar molecules) theories;41–43 exceptions are reactions which are close to thermoneutral and those of bulky cations with C2F4. In many reactions of ions with intermediate RE values (ca. 12–16 eV), thermochemistry can indicate which neutral product(s) form with the product cation. Many of the reactions of ions with high RE (i.e. greater than ca. 14 eV) probably proceed via electron transfer. However, the reactions of both N+ and N2+ with C2H4 give very different products from their reactions with the four fluorinated ethenes; the former reactions can only form some of the observed products if a chemical intermediate forms between Nx+ and C2H4. Energetics cannot be the only factor in determining the outcome of these IM reactions, because one might then expect the reactions of CO+ and Kr+, with near identical RE values, to produce similar products with a common fluoroethene; that is not observed. Finally, it is difficult to determine whether the presence or absence of a FC gap of the fluorinated ethene at the RE of the reactant ion is a significant factor in determining whether electron transfer is the dominant mechanism. It was noted earlier that electron transfer can either take place at ‘long range’ where one might expect FC vibrational overlap factors to be a significant factor in determining the efficiency of the reaction, or at ‘short range’ where FC factors may be perturbed by changes to the potential energy surfaces of the reacting species as they interact with each other.3 Thus, reactions do occur by electron transfer in which the FC factor of the neutral reactant with a vibronic state of its parent ion can be both significant and not significant.
When comparing the product state distributions from the chemical IM and photoionisation reactions, again and disappointingly no clear pattern emerges. There are many reactions where good agreement is observed between the two sets of branching ratios, suggesting that either long- or short-range electron transfer is the dominant mechanism for the chemical reaction. There is some evidence that the agreement deteriorates as the number of fluorine atoms in C2HxF4−x increases. So long as the RE of the cation exceeds the IE of the fluoroethene, there is no clear evidence whether energetics is a major factor in determining agreement or otherwise between branching ratios; for example, for atomic rare gas ions, Ne+ (RE = 21.56 eV) and Xe+ (12.13 eV) give reasonable agreement, but Kr+ with an intermediate RE of 14.00 eV gives poor agreement. There is some suggestion, however, that the greater the excess energy between the RE(ion) and the IE(fluoroethene) the better the agreement, so it may be that the reactions of Kr+ are anomalous. Finally, one point keeps occurring in such comparisons,23,24,36,37 observed most clearly in the reactions of the chlorinated ethenes;24 that when disagreements are significant between the two sets of BRs, then the parent ion and larger daughter ions are more prevalent in the chemical IM reactions, whereas the smaller daughter ions are more prevalent in the photoionisation reaction. One partial explanation may be that the full recombination energy is not available in the IM reaction of a molecular ion because the neutralised ion may form internally excited. However, there are significant discrepancies with atomic ions which cannot be explained by this factor.
It is only with the advent of vacuum-UV beamlines located on 3rd generation very stable synchrotron sources operating in ‘top-up’ mode that one is able to collect ion breakdown diagrams of polyatomic molecules via coincidence spectroscopy with signal-to-noise ratios so good that it is possible to make these kinds of comparison; individual photoionisation BRs are accurate to ca. ±1% in this work at the Swiss Light Source,35,60 to be compared with ca. 10% in our earlier studies at the 2nd generation Daresbury synchrotron source.23,24,36,37 From a fundamental perspective, the challenge now is to improve the accuracy of IM reactions that their product BRs can be determined with a much improved accuracy than the ±20% that is normal for such current studies.
Acknowledgements
We thank Mr Ben Hopkins for help in collecting some of the data. Thermochemical calculations were performed using recent data on the four fluorinated ethene molecules obtained by Drs Jonelle Harvey and Andras Bodi at the Swiss Light Source.33,35 This paper presents results from experiments taken in the Molecular Physics Group Laboratory of the School of Physics and Astronomy, University of Birmingham proposed by and under the supervision of Dr Christopher A. Mayhew. We are particularly grateful to him for pointing out to us the significance of ref. 49–51. The work was funded by EPSRC (EP/E027571/1 and EP/E00914X/1).References
- United Nations Environment Protocol, Montreal Protocol, 1987, http://ozone.unep.org/Ratification_status/montreal_protocol.shtml.
- UN Inter-Governmental Panel on Climate Change, Working Group I, 4th Assessment Report, 2007, ch. 1 and 2.
- G. K. Jarvis, C. A. Mayhew, R. A. Kennedy and R. P. Tuckett, Int. J. Mass Spectrom., 2000, 202, 323 CrossRef CAS.
- M. J. McEwan, G. B. I. Scott and V. G. Anicich, Int. J. Mass Spectrom. Ion Processes, 1998, 172, 209 CrossRef CAS.
- V. G. Anicich and M. J. McEwan, Planet. Space Sci., 1997, 45, 897 CrossRef CAS.
- K. K. Matthews, N. G. Adams and N. D. Fisher, J. Phys. Chem. A, 1997, 101, 2841 CrossRef CAS.
- D. A. Fairley, G. B. I. Scott, C. G. Freeman, G. A. R. Maclagan and M. J. McEwan, J. Phys. Chem. A, 1997, 101, 2848 CrossRef CAS.
- G. K. Jarvis, C. A. Mayhew and R. P. Tuckett, J. Phys. Chem., 1996, 100, 17166 CrossRef CAS.
- R. A. Morris, A. A. Viggiano and J. F. Paulson, J. Phys. Chem., 1993, 97, 6208 CrossRef CAS.
- M. Tsuji, H. Kouno, K. Matsumura, T. Funatsu, Y. Nishimura, H. Obase, H. Kugishima and K. Yoshida, J. Chem. Phys., 1993, 98, 2011 CrossRef CAS PubMed.
- D. Smith, P. Spanel and C. A. Mayhew, Int. J. Mass Spectrom. Ion Processes, 1992, 117, 457 CrossRef CAS.
- C. A. Mayhew and D. Smith, J. Phys. B: At., Mol. Opt. Phys., 1990, 23, 3139 CrossRef CAS.
- C. A. Mayhew and D. Smith, Int. J. Mass Spectrom. Ion Processes, 1990, 100, 737 CrossRef CAS.
- K. Giles, N. G. Adams and D. Smith, J. Phys. B: At., Mol. Opt. Phys., 1989, 22, 873 CrossRef CAS.
- A. B. Rakshit, Int. J. Mass Spectrom. Ion Processes, 1986, 69, 45 CrossRef.
- D. K. Bohme and G. I. Mackay, J. Am. Chem. Soc., 1981, 103, 2173 CrossRef CAS.
- D. Smith and N. G. Adams, Chem. Phys. Lett., 1980, 76, 418 CrossRef CAS.
- A. B. Rakshit, Z. Naturforsch., A: Phys., Phys. Chem., Kosmophys., 1980, 35, 1218 Search PubMed.
- A. B. Rakshit and P. Warneck, J. Chem. Soc., Faraday Trans. 2, 1980, 76, 1084 RSC.
- A. B. Rakshit and N. D. Twiddy, Chem. Phys. Lett., 1979, 60, 400 CrossRef CAS.
- G. A. W. Derwish, A. Galli, A. Giardini-Guidoni and G. G. Volpi, J. Am. Chem. Soc., 1964, 86, 4563 CrossRef CAS.
- V. G. Anicich, NASA JPL Report 03-19, 2003.
- V. Mikhailov, M. A. Parkes, R. P. Tuckett and C. A. Mayhew, J. Phys. Chem. A, 2006, 110, 5760 CrossRef CAS PubMed.
- V. Mikhailov, M. A. Parkes, M. J. Simpson, R. P. Tuckett and C. A. Mayhew, J. Phys. Chem. A, 2008, 112, 9012 CrossRef CAS PubMed.
- M. J. Simpson and R. P. Tuckett, J. Phys. Chem. A, 2012, 116, 8119 CrossRef CAS PubMed.
- S. G. Lias, J. E. Bartmess, J. F. Liebman, J. L. Holmes, R. D. Levin and W. G. Mallard, J. Phys. Chem. Ref. Data, 1988, 17(suppl 1) CAS.
- R. M. O'Malley, K. R. Jennings, M. T. Bowers and V. G. Anicich, Int. J. Mass Spectrom. Ion Phys., 1973, 11, 89 CrossRef CAS.
- A. J. Ferrer-Correia and K. R. Jennings, Int. J. Mass Spectrom. Ion Phys., 1973, 11, 111 CrossRef CAS.
- V. G. Anicich, M. T. Bowers, R. M. O'Malley and K. R. Jennings, Int. J. Mass Spectrom. Ion Phys., 1973, 11, 99 CrossRef CAS.
- V. G. Anicich and M. T. Bowers, Int. J. Mass Spectrom. Ion Phys., 1974, 13, 351 CrossRef CAS.
- V. G. Anicich and M. T. Bowers, Int. J. Mass Spectrom. Ion Phys., 1974, 13, 359 CrossRef CAS.
- B. A. Williams and T. A. Cool, J. Chem. Phys., 1991, 94, 6358 CrossRef CAS PubMed.
- J. Harvey, P. Hemberger, A. Bodi and R. P. Tuckett, J. Chem. Phys., 2013, 138, 124301 CrossRef PubMed.
- C. R. Brundle, M. B. Robin, N. A. Kuebler and B. Harold, J. Am. Chem. Soc., 1972, 94, 1451 CrossRef CAS.
- J. Harvey, A. Bodi, R. P. Tuckett and B. Sztaray, Phys. Chem. Chem. Phys., 2012, 14, 3935 RSC.
- M. A. Parkes, R. Y. L. Chim, C. A. Mayhew, V. Mikhailov and R. P. Tuckett, Mol. Phys., 2006, 104, 263 CrossRef CAS.
- C. R. Howle, D. J. Collins, R. P. Tuckett and A. E. R. Malins, Phys. Chem. Chem. Phys., 2005, 7, 2287 RSC.
- D. Smith and N. G. Adams, Adv. At. Mol. Phys., 1988, 24, 1 CAS.
- M. J. Simpson, PhD thesis, University of Birmingham, 2010, http://etheses.bham.ac.uk/1056/.
- M. W. Chase, J. Phys. Chem. Ref. Data, Monogr., 1998, 9 Search PubMed.
- P. M. Langevin, Ann. Chim. Phys., 1905, 5, 245 CAS.
- G. Gioumousis and D. P. Stevenson, J. Chem. Phys., 1958, 29, 294 CrossRef CAS PubMed.
- T. Su and W. J. Chesnavich, J. Chem. Phys., 1982, 76, 5183 CrossRef CAS PubMed.
- D. R. Lide, Handbook of Chemistry and Physics, Taylor and Francis, London UK, 89th edn, 2008 Search PubMed.
- K. J. Miller, J. Am. Chem. Soc., 1990, 112, 8533 CrossRef CAS.
- R. Stockbauer and M. G. Inghram, J. Chem. Phys., 1975, 62, 4862 CrossRef CAS PubMed.
- F. Innocenti, M. Eypper, E. P. F. Lee, D. K. Stranges, D. K. W. Mok, F. Chau, G. C. King and J. M. Dyke, Chem.–Eur. J., 2008, 14, 11452 CrossRef CAS PubMed.
- S. Borkar and B. Sztaray, J. Phys. Chem. A, 2010, 114, 6117 CrossRef CAS PubMed.
- G. Nicol, J. Sunner and P. Kebarle, Int. J. Mass Spectrom. Ion Processes, 1988, 84, 135 CrossRef CAS.
- D. K. Bohme, G. I. Mackay and H. I. Schiff, J. Chem. Phys., 1980, 73, 4976 CrossRef CAS PubMed.
- G. Bouchoux, J. Y. Salpin and D. Leblanc, Int. J. Mass Spectrom. Ion Processes, 1996, 153, 37 CrossRef CAS.
- W. Lindinger, A. Hansel and A. Jordan, Int. J. Mass Spectrom., 1998, 173, 191 CrossRef CAS.
- A. Bodi, M. Johnson, T. Gerber, Z. Gengeliczki, B. Sztaray and T. Baer, Rev. Sci. Instrum., 2009, 80, 034101 CrossRef PubMed.
- R. Bombach, J. Dannacher and J. P. Stadelmann, Int. J. Mass Spectrom. Ion Processes, 1984, 58, 217 CrossRef CAS.
- J. W. Gallagher, C. E. Brion, J. A. R. Samson and P. W. Langhoff, J. Phys. Chem. Ref. Data, 1988, 17, 9 CrossRef CAS PubMed.
- J. Harvey, private communication of unpublished data, 2013.
- J. E. Pollard, D. J. Trevor, R. E. Reutt, Y. T. Lee and D. A. Shirley, J. Chem. Phys., 1984, 81, 5302 CrossRef CAS PubMed.
- S. Eden, P. Limao-Vieira, P. A. Kendall, N. J. Mason, J. Delwiche, M. J. Hubin-Franskin, T. Tanaka, M. Kitajima, H. Tanaka, H. Cho and S. V. Hoffmann, Chem. Phys., 2004, 297, 257 CrossRef CAS PubMed.
- C. E. Moore, Atomic Energy Levels, National Bureau of Standards, NSRDS-NBS 35, vol. I, re-issued 1971.
- J. Harvey, R. P. Tuckett and A. Bodi, J. Phys. Chem. A, 2012, 116, 9696 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp54881c |
| ‡ Current address: Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. |
| § Current address: King Edwards High School for Girls, Edgbaston Park Road, Birmingham B15 2UB, UK. |
| ¶ Current address: Physical and Theoretical Chemical Laboratories, South Parks Road, Oxford OX1 3QZ, UK. |
| This journal is © the Owner Societies 2014 |