 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rate of gas absorption on a slippery bubble mattress
Elif
Karatay
,
Peichun Amy
Tsai
and
Rob G. H.
Lammertink
*
Soft Matter, Fluidics and Interfaces, MESA+ Institute for Nanotechnology, University of Twente, 7500AE, Enschede, The Netherlands. E-mail: r.g.h.lammertink@utwente.nl; Fax: +31 53 489 2882; Tel: +31 53 489 4798
First published on 1st October 2013
Abstract
We investigate the absorption of a pure gas into a liquid in laminar flow past a superhydrophobic surface consisting of alternating solid walls and micro-bubbles. We experimentally measure and numerically estimate the dynamic mass transfer of gas absorption at stable gas–liquid interfaces for short contacting times. We study the net rate of gas absorption experimentally by in situ measurements of dissolved oxygen concentration profiles in aqueous solutions flowing over oxygen bubbles by fluorescence lifetime imaging microscopy. We numerically analyze the dynamics of interfacial mass transfer of dissolved oxygen by considering (i) kinetic equilibrium conditions at bubble surfaces that are conventionally described by Henry's Law and (ii) non-equilibrium conditions at bubble surfaces using Statistical Rate Theory (SRT). Our experimental results show that kinetic equilibrium is not established for short contact times. Mass transfer of gas into liquid flow past micro-bubbles can be well described by our simulations performed with the non-equilibrium theory for a short exposure time (∼180 μs) of liquid with a microbubble, deviating from the commonly accepted Henry's Law.
1 Introduction
Gassing/degassing of liquids is of interest in various research fields and applications ranging from the multiphase chemical reactors to biotechnologies in both the macro- and micro-scale.1–3 Traditional gas–liquid contacting equipment such as distillation columns, packed towers, tray/plate columns and bubble columns provide direct contact of gas–liquid and phase equilibrium-based absorption/desorption processes at these interfaces. The mass transfer in such processes has been studied extensively and the mass transfer resistances have been reported to be determined by the transport of gaseous and dissolved species in the gas and liquid phases, respectively.1,4,5 Under equilibrium conditions, the concentrations in both gas and liquid phases at the gas–liquid boundary are proportional to their partial pressures, which are commonly described by Henry's Law.6Besides the conventional gas–liquid contacting equipment, porous hydrophobic membranes in gas–liquid contacts are increasingly utilized because of several advantages, such as their fixed interfacial area used for the stabilization of the gas–liquid interfaces.7–10 Mass transfer characteristics of gas absorption/desorption in membrane contactors have been widely studied.7,10–15 The overall mass transfer process is reported to consist of four consecutive steps: (i) transport from the bulk gas phase to the outer surface of the membrane, (ii) diffusion through the membrane, (iii) dissolution of gas into liquid, and (iv) liquid phase transport. In these studies, the overall mass transfer coefficients are investigated. Analytical and experimental mass transfer correlations are obtained for developing/fully developed boundary layers using analogous heat transfer solutions of the Graetz–Lévèque type.14,15 The mass transfer resistance in the liquid phase, in relation to operating parameters, and the mass transfer resistance in the membrane, in relation to the wetting phenomena, have been investigated in detail.12,13,15 The mass transfer resistances in the gas phase and at the gas–liquid surfaces are commonly neglected due to the fast gas phase diffusion and the assumption of equilibrium at the gas–liquid boundaries.
The gas dissolution at the phase boundary, however, has been reported to induce a major mass transfer resistance, often determining or limiting the performance of gas absorption/desorption processes.16–25 The surface resistances have been investigated in many theoretical studies using both phase-equilibrium and non-equilibrium conditions. For example, the stagnant film theory,18 Higbie's penetration theory19 and the film-penetration theory20 are among the widely used interface models under equilibrium conditions. The dynamics of non-equilibrium interface transport has also been investigated in previous analytical and theoretical studies based on kinetic and thermodynamic considerations.22,24 Moreover, the mass transfer resistance of gas dissolution at the gas–liquid interfaces has been reported as the rate limiting step in gas absorption/desorption processes for short contacting times.22,25,26 The non-equilibrium based interface models have been employed in various experimental studies investigating the evaporation of suspended droplets, the gas bubble evolution in liquids and gas adsorption/desorption kinetics on catalytic systems.25,27–31
However, to the best of our knowledge, the mass transfer resistances of gas dissolution into a liquid under pressure-driven laminar flow past slippery hydrophobic substrates with a slip velocity have not been investigated. To generate hydrodynamic slippage, hydrophobic substrates containing gas bubbles have been used in recent studies,32–34 as the liquid flows over those shear-free bubble surfaces. Effective slip lengths – quantifying the slippage – in the micrometer range are obtained for laminar flow past superhydrophobic substrates in both experimental and theoretical studies.32–40 Such fluidic platforms with slippery interfaces have been shown to enhance liquid convection and have been suggested to increase the interfacial mass transport rates.33,39,41 In a recent theoretical study,42 enhancements of up to two orders of magnitude in interfacially driven transport phenomena are predicted on slippery surfaces. These studies highlight the room for investigation of interfacial gas transport at bubble surfaces. In our previous study,33 we presented a hydrophobic microfluidic device that allows for the formation of stable and active – gas pressure controlled – micro-bubbles at the boundary of microchannels. We experimentally and numerically examined the effect of micro-bubble protrusion angle θ and surface porosity on the slippage on our bubble mattresses. Our results revealed the dependency of hydrodynamic slip on the gas–liquid interface curvature and surface porosity.
In this study, we examine experimentally and numerically the oxygen absorption into a water stream in a pressure-driven flow over a superhydrophobic surface with transversely embedded oxygen gas micro-bubbles (Fig. 1). The fabricated devices allow for tunable gas phase pressure, bubble geometry, and liquid phase flow.33 We measure the detailed spatial dissolved oxygen gas concentration profiles in the aqueous phase with frequency-domain fluorescence lifetime imaging microscopy (FLIM). We numerically study the dynamics of interfacial mass transfer of dissolved oxygen in the liquid side microchannel embedded with curved oxygen microbubbles, using two models of interfacial gas concentration at the (i) kinetic equilibrium state and (ii) non-equilibrium state using Statistical Rate Theory (SRT).
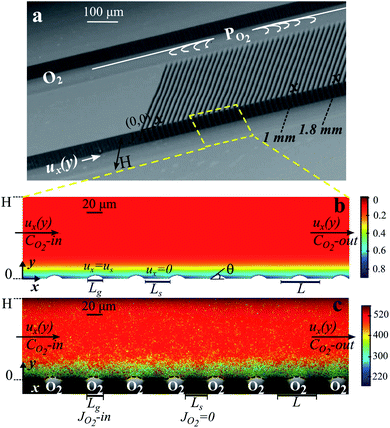 | ||
| Fig. 1 Microfluidic bubble mattress and oxygen dissolution at bubble surfaces. (a) Scanning electron microscopy image of the microfluidic device showing two main microchannels for oxygen gas (PO2) and water (ux(y)) streams connected by oxygen-filled side channels. (b) Numerical results of the dissolved oxygen concentration in the water side microchannel solved with identical operating settings as in (c). (c) Lifetime field resolved by FLIM superimposed on the bright-field microscopy image showing bubbles protruding at 43° ± 2° into the water side microchannel with a height of H = 100 μm. Lg is the width of the oxygen-filled side channel (Lg = 20 μm), and Ls is the width of the solid boundary (Ls = 32.6 μm). The color bars refer to the lifetime of RTDP aqueous solution which is given in ns (c) and the oxygen concentration which is given in mol m−3 in (b). In b and c, surface porosity φ = 0.38 and Qw = 45 μl min−1. | ||
2 Experimental methods
2.1 Microfluidic setup
Fig. 1a shows a representative scanning microscopy image of silicon-glass based microfluidic devices. The microfluidic devices consist of two main parallel microchannels for separate aqueous solution and oxygen gas streams, connected by an array of side channels.33 Silicon microchannels were fabricated by standard photolithography followed by a deep reactive ion etching process. The microchannels were sealed by anodic bonding to glass. To prevent the wetting of the connecting side channels, the original hydrophilic silicon microchannels were hydrophobized. We adopted the protocol described in ref. 43 for the hydrophobization via ultraviolet (UV) light illumination of silicone oil. The width of the main microchannels H was 50 μm or 100 μm. The width of the gas-filled side channels Lg was kept constant at 20 μm and the length of the liquid–solid interface Ls was 20 μm or 30 μm. The periodic bubble unit length is defined as L = Lg + Ls and the surface porosity (shear-free fraction, φ = Lg/L) is 0.38 or 0.54 for different Ls (Fig. 1). The depth of the microchannels was 100 μm.The flow rate of the liquid side was controlled using a syringe pump (HARVARD Apparatus PHD 2000) and the pressure of the oxygen gas stream was controlled using a pressure controller (Bronkhorst, EL-PRESS, P-602C). The side-channels were filled with oxygen gas due to the hydrophobicity of the microchannel walls and sufficiently large gas pressure Pg. The protrusion angles θ of the oxygen micro-bubbles into the aqueous stream were controlled by the active control of the applied oxygen gas pressure. The protrusion angles of the oxygen micro-bubbles were varied in a range from 2° to 45° by changing the applied gas pressure (∼0.6 to ∼1.2 bar) for varying liquid flow rates from 9 μl min−1 to 45 μl min−1. During the measurements the micro-bubbles were stable and symmetrically pinned at the sharp corners of the side channels.
2.2 FLIM experiments
The local oxygen concentrations dissolved in the liquid side channels were quantified by using a Ru-based oxygen sensitive luminescent dye, ruthenium tris(2,2′-dipyridyl) dichloride hexahydrate (RTDP) (Sigma Aldrich). The frequency domain fluorescence lifetime imaging microscopy (FD-FLIM) technique was employed to measure the lifetime domains of RTDP aqueous solutions in the microchannels.44,45 With the lifetime measurements, the dissolved oxygen concentrations can be calculated.44 For the FLIM experiments, a LIFA-X system (Lambert Instruments) was used on a Zeiss Axio Observer inverted microscope attached to a modulated LED light source and an intensified CCD camera (LI2CAM) in gated mode. During the measurements, the intensifier MCP (micro-channel plate) voltage was kept at 650 V. The optical sensing was carried out at a fixed modulation frequency of 100 kHz. We used 10 μM fluorescein (Sigma Aldrich) solution as a reference fluorophore to test any instrumentation phase shift at the operating frequency. Fluorescein is suitable as a reference phase shift fluorophore due to its short lifetime τ < 6 ns compared to RTDP (∼600 ns). The data acquired were analyzed with the LI-FLIM software package to resolve the lifetime fields.400 μM aqueous solutions of RTDP were prepared. Lifetimes of the RTDP were measured in oxygen-free (N2 saturated), aerated and oxygen-saturated aqueous solutions. The solutions were saturated with a certain gas by bubbling that gas through the solution for sufficiently long times. The dissolved oxygen contents of the solutions were confirmed using a fiber optic oxygen sensor (FIBOX 3, PreSens). For these measurements using the microfluidic platform, the microbubbles were established by nitrogen, air and oxygen gases for corresponding oxygen-free, aerated and oxygen-saturated conditions.
During the mass transfer experiments, oxygen gas microbubbles were established at the boundary of the microchannels (Fig. 1c and 3). If otherwise not stated, deoxygenated RTDP aqueous solution was the working liquid flowing past the transversely aligned oxygen bubbles. The experiments were performed at room temperature without further temperature regulation.
3 Numerical simulations
3.1 Governing equations
Numerically we study the mass transfer characteristics of oxygen gas dissolution at the surfaces of curved oxygen microbubbles aligned perpendicular to a pressure-driven microfluidic laminar flow. We solve for coupled mass and momentum transport on our bubble mattress geometry (Comsol Multiphysics v4.3). The computational domain represents the experimental geometric parameters (Fig. 1b) and we solve for velocity and concentration profiles in the liquid side microchannels with the same experimental operating settings. In our simulations, the gas side transport is not considered since the effects of mass transfer limitations in the gas phase and the effects of oxygen depletion during gas dissolution vanish due to the active supply of pure oxygen gas.The Navier–Stokes equations and the conservation of species equation (convection-diffusion) are solved for a steady pressure-driven flow of water in a microchannel consisting of 60 successive bubble units at the bottom surface (y = 0). In order to eliminate any developing flow effects and entrance/outlet effects, an entrance and outlet section of 2-bubble unit cell length was included in the front and end of the bubble mattress. The bubbles are non-deformed and pinned at the corners of side channels, as observed in the experiments. We approximate the bubble profiles as circular arcs. We parametrize the bubble interface curvature by the protrusion angle θ (Fig. 1b).
The pressure-driven laminar flow was produced by applying a mean velocity as the inlet condition. We varied the inlet mean velocities in the range of 0.04 to 0.2 m s−1, consistent with our experiments. We applied shear-free boundary conditions (i.e. perfect slip) for the bubble surfaces and no-slip boundary conditions for the solid microchannel walls (at the bottom wall along each Ls at y = 0 and the upper wall at y = H). The convective velocity (u) was obtained from the Navier–Stokes equations.
The steady-state conservation of species eqn (1) was solved for the dissolved oxygen transport. The oxygen dissolves at the microbubble surfaces.
| ∇·(uCO2) = ∇·(D∇CO2) | (1) |
Here D is the diffusion coefficient of oxygen in water (= 2.67 × 10−9 m2 s−1 at 25 °C (ref. 25)). CO2 is the dissolved oxygen concentration in water. The inlet boundary condition to the microchannel was set at a constant concentration value. If otherwise not stated, the inlet concentration is CO2|(x=0) = 0, representing deoxygenated solutions in the experiments. We applied no-flux boundary conditions (∂CO2/∂y = 0) at the solid microchannel walls (at each Ls at y = 0 and the upper wall at y = H), as the silicon microchannel walls are not permeable to oxygen.
The mass transfer boundary conditions at the bubble surfaces represent the gas dissolution mechanism at the gas–liquid interface that determines the rate of gas absorption by the liquid. We examine two different boundary conditions along the gas protrusions as described below (Fig. 2).
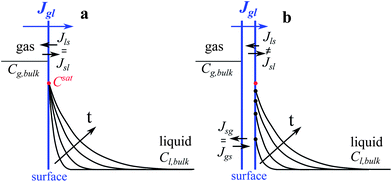 | ||
Fig. 2 Schematic illustration of the applied boundary conditions and the effects of interface resistances on the concentration profiles in the presence of liquid flow. (a) The phase equilibrium state where the interface liquid concentration is fixed at the saturation value given by Henry's Law ( ). The time (t) evolution results in increasing boundary layer thicknesses. (b) The non-equilibrium state where the interface liquid concentrations ( ). The time (t) evolution results in increasing boundary layer thicknesses. (b) The non-equilibrium state where the interface liquid concentrations ( ) are determined by the instantaneous exchange rates of absorbed and desorbed molecules at the surface liquid boundary (sl). The time (t) evolution results in increasing boundary layer thicknesses and increasing interface concentrations approaching the saturation value. In (a and b) the mass transfer resistances in the bulk gas phase are neglected. ) are determined by the instantaneous exchange rates of absorbed and desorbed molecules at the surface liquid boundary (sl). The time (t) evolution results in increasing boundary layer thicknesses and increasing interface concentrations approaching the saturation value. In (a and b) the mass transfer resistances in the bulk gas phase are neglected. | ||
3.2 Mass transfer boundary conditions
The mass transfer resistance across a gas–liquid interface is investigated in many previous theoretical studies.16–25 The surface resistances are commonly described under phase-equilibrium conditions. Higbie's penetration theory19 and the two film-penetration theory20 are among the well-known mathematical models. It is important to note that these theories consider equilibrium conditions at the interface. The net exchange rate of absorbed and desorbed species at the surface determines the amount of accumulated species at the interface and thereby the interface concentration. Under equilibrium conditions, the amount of accumulated species is given by Henry's Law.6The mass transfer resistance at the interfaces has been investigated also for dynamical non-equilibrium conditions in previous theoretical and analytical studies using kinetic and thermodynamic approaches.22–25 In kinetic interface models, the net exchange rate at the interface is described by the Hertz–Knudsen equation where mass accommodation (condensation) coefficients are used as fitting parameters.22,23
Besides the kinetic approaches, a statistical rate theory (SRT) is suggested to predict the instantaneous rate of interfacial molecular transport.24,25 In SRT the gas–liquid boundary is described to consist of two interfaces; the gas–surface interface and the surface–liquid interface, with the consideration of the Gibbs dividing surface26 (Fig. 2b). The molecular distributions in each phase (bulk gas, surface, and bulk liquid) are related to thermodynamic properties using the Boltzmann definition of entropy. The transition probability of molecules from one phase to another is derived via a first order perturbation analysis of the Schrödinger equation. The calculated transition probability of gaseous species from the bulk gas phase to the bulk liquid phase has been used in approximating the instantaneous non-equilibrium exchange rate of absorbed and desorbed molecules at the surfaces. For long contacting times, the SRT predicts equilibrium exchange rates and equilibrium interface concentrations at the saturation value.26
The theoretical models16–25 for interfaces would suggest that on hybrid substrates with hydrodynamic slippage, the mass transfer resistance of gas dissolution at the phase boundary can become the rate limiting step for short contacting times of the liquid with bubbles. Therefore in our numerical simulations, in addition to the phase equilibrium state (Fig. 2a), we also study the dynamics of interfacial mass transfer at curved oxygen microbubbles at the non-equilibrium state (Fig. 2b).
Firstly, the saturation concentration of oxygen in water is applied as boundary conditions at the bubble surfaces in our phase-equilibrium based simulations (Fig. 2a). Under equilibrium conditions, the saturation value of the dissolved oxygen concentration at the interface 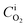 is proportional to its partial pressure PO2 and described by Henry's Law6
is proportional to its partial pressure PO2 and described by Henry's Law6
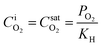 | (2) |
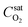 is the saturation concentration of oxygen in water. For pure oxygen bubbles, the dissolved oxygen concentration at the interface is calculated to be
is the saturation concentration of oxygen in water. For pure oxygen bubbles, the dissolved oxygen concentration at the interface is calculated to be 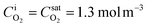 at 1 atm and 25 °C.
at 1 atm and 25 °C.
Secondly, the rate of molecular transport between the surface phase and the bulk liquid phase is predicted by SRT and applied as flux boundary conditions along the gas protrusions (coinciding each Lg at y = 0) in our non-equilibrium based simulations. According to SRT, this instantaneous molecular rate of transport between the phases is described as:
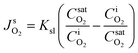 | (3) |
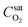 is the fixed equilibrium concentration of the dissolved oxygen, hence the solubility at the given temperature and pressure.
is the fixed equilibrium concentration of the dissolved oxygen, hence the solubility at the given temperature and pressure. 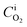 is the interface concentration of oxygen in the liquid phase. At the bubble surface, concentration is a function of the axial x location and to some extent the bubble protrusion depth into liquid in the y direction (Fig. 2). Ksl (moles m−2 s−1) is the equilibrium oxygen exchange rate between the bubble surface and the liquid phase and has a constant value for a system with given local thermodynamic properties.
is the interface concentration of oxygen in the liquid phase. At the bubble surface, concentration is a function of the axial x location and to some extent the bubble protrusion depth into liquid in the y direction (Fig. 2). Ksl (moles m−2 s−1) is the equilibrium oxygen exchange rate between the bubble surface and the liquid phase and has a constant value for a system with given local thermodynamic properties.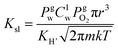 | (4) |
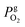 is the partial pressure of oxygen in the gaseous phase and is approximated by neglecting the Laplace pressure as
is the partial pressure of oxygen in the gaseous phase and is approximated by neglecting the Laplace pressure as 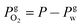 where P is the total pressure at the bubble surface. Clw is the concentration of water in the liquid phase, which is 55.2 M under the experimental conditions. Here, r and m are the radius and molecular mass of the oxygen gas molecule, k is the Boltzmann constant, and T is the temperature. KH′ is the “pressure-based” Henry's law constant, given as 4.85 × 109 Pa at 25 °C.1
where P is the total pressure at the bubble surface. Clw is the concentration of water in the liquid phase, which is 55.2 M under the experimental conditions. Here, r and m are the radius and molecular mass of the oxygen gas molecule, k is the Boltzmann constant, and T is the temperature. KH′ is the “pressure-based” Henry's law constant, given as 4.85 × 109 Pa at 25 °C.1
Without any fitting parameters, with a known value of Ksl, we implemented the expression given in eqn (3) as boundary conditions defining the oxygen flux at the bubble surfaces. It is important to note here that for longer contacting times, the interface concentration approaches the saturation value 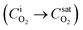 , and the rate of transport between phases
, and the rate of transport between phases 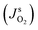 vanishes, which is indicative of equilibrium conditions, i.e. a constant concentration, at the bubble surface.
vanishes, which is indicative of equilibrium conditions, i.e. a constant concentration, at the bubble surface.
4 Results and discussion
4.1 FLIM measurements
Oxygen has been reported elsewhere46 to be an effective collisional dynamic quencher of RTDP and its effect on RTDP fluorescence decay is described by the Stern–Volmer equation (5)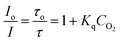 | (5) |
4.2 Rate of O2 absorption
During the mass transfer experiments, oxygen gas microbubbles were established at the boundary of the microchannels (Fig. 3). We measured the lifetime of RTDP across the liquid side microchannel height (0 ≤ y ≤ H) at different axial locations x (Fig. 3). The bubble interface profiles and locations were experimentally determined by locating the minimum lifetime data measured near the hybrid wall. The bubble profiles were also calculated by circular arc estimation and depicted by the dashed lines in Fig. 3. Fig. 3 shows the successive lifetime fields measured with FLIM at different axial locations along the same microchannel embedded with microbubbles θ = 43° ± 2°. In Fig. 3c, the thickness of the diffusion boundary layer is ∼23% of the microchannel height H, and does not extend further into the microchannel due to a relatively large Reynolds number Re = 7.5.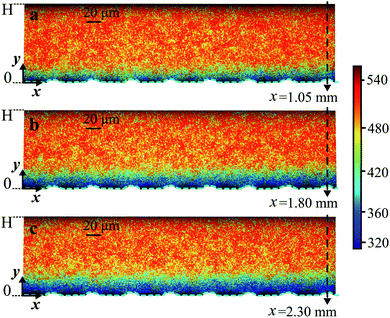 | ||
| Fig. 3 Successive lifetime fields in axial position x. Quantitative visualization of the increasing boundary layer thickness along downstream flow with Qw = 45 μl min−1 at (a) 0.7 ≲ x ≲ 1.1, (b) 1.4 ≲ x ≲ 1.8, and (c) 1.9 ≲ x ≲ 2.3. Here θ = 43° ± 2° and φ = 0.38. In (a–c) the flow direction is from left to right. The color bar refers to the lifetime, which is given in nanoseconds. The dashed arrows indicate the axial positions at which the local oxygen concentration profiles across the microchannel height are obtained. | ||
From the lifetime fields as shown in Fig. 3, the dissolved oxygen concentration gradients were obtained by correlating the lifetime data by eqn (5). When processing the data, no filtering or smoothing was applied to the measured lifetime fields. Due to the scattering of the experimental lifetime data, the obtained oxygen concentration values were averaged over the bubble surfaces (over ∼30 pixels in x, equivalent to Lg). The presented results are the average of independent experiments performed with the same operating fluidic settings. The statistical error bars are calculated from these experiments.
Fig. 4a–c represent the local dissolved oxygen concentration profiles obtained from the lifetime fields shown in Fig. 3a–c, which are successive measurements at different axial x positions in the microchannel during the same experiment. The local profiles across the microchannel height, with respect to the y direction normal to the bubble surfaces, are obtained at x positions coinciding with the bubble surfaces that are indicated with dashed arrows in Fig. 3. Here the deoxygenated RTDP aqueous solution flows at a Reynolds number Re = 7.5. The experimental results are compared with numerical results obtained for phase-equilibrium based and SRT-based gas absorption.
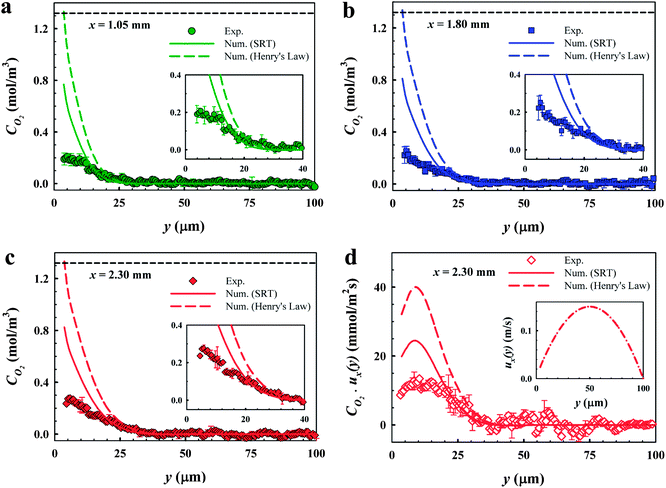 | ||
| Fig. 4 Successive dissolved oxygen profiles at different axial x positions. (a) Local O2 profile at x = 1.05 mm. The inset is a zoom-in of the O2 profile in (a). (b) Local O2 profile at x = 1.80 mm. The inset is a zoom-in of the O2 profile in (b). (c) Local O2 profile at x = 2.30 mm. The inset is a zoom-in of the O2 profile in (c). (d) Local convective flux JO2 profile at x = 2.30 mm. The inset shows the flow velocity profile obtained by simulations performed at the same experimental parameters. In (a–d) symbols represent the experimental results obtained by FLIM, the color solid lines represent the numerical results solved with SRT, and the color dashed lines represent the numerical results solved using Henry's law. Here the microchannel height H = 100 μm, θ = 43° ± 2°, φ = 0.38, and Re = 7.5. The black dashed lines in (a–c) depict the equilibrium saturation value of oxygen in water. | ||
The bubble interface position is y ∼ 3.8 μm in Fig. 4 due to a large protrusion angle θ = 43° ± 2° of the bubbles into the water stream. The interface O2 concentration predicted by phase-equilibrium based simulations converges to the saturation value 1.3 mol m−3 as imposed by Henry's law. Whereas the interfacial dissolved gas concentration predicted by SRT based simulations is ∼0.8 mol m−3 at x = 1.05 mm and slightly increases for increasing x. When y ≳ 3.8 μm, the oxygen concentration gradients are steep due to a relatively large Reynolds number. As can be clearly seen in Fig. 4a–c, the effect of the microbubbles on the concentration profiles is damped when y ≳ 25 μm at x = 1.05 mm and when y ≳ 35 μm at x = 2.30 mm. Beyond the boundary layers, the oxygen concentration drops to the inlet bulk concentration value. Our findings are consistent with previous studies45,51 reporting the local oxygen gradients resolved by in situ FLIM measurements. In these studies, oxygen diffuses from an oxygen enriched liquid phase to an oxygen-free liquid phase.
The FLIM measurements in Fig. 4a–c reveal very low concentrations near bubble interfaces when y ≲ 10 μm. There is a sharp discrepancy between the measurements and both numerical results in the proximity of the bubble interfaces (3.8 μm ≤ y ≲ 10 μm). The standard errors calculated close to the bubble surface are also larger compared to those of the remainder of the microchannel. The deviation can be related to the systematic error in our FLIM measurements close to the gas–liquid interfaces (additional measurements are provided in the Appendix). The deviations near the bubble surfaces do not interfere with the results presented here since we do not consider the experimental data obtained in the proximity of bubble interfaces.
Considering the oxygen concentration data when ≈10 μm ≤ y ≤ H, the measured oxygen concentration profiles are in good agreement with the simulations performed with an incorporated SRT boundary condition. Whereas, the numerical results computed for a constant interface concentration at the solubility limit of oxygen in water predict steeper profiles and faster gas absorption than those in the measurements. Due to a relatively large Re = 7.5, the water residence time within the hold-up volume of the microchannel embedded with the bubbles is as small as 96 ms. The inset in Fig. 4d shows the numerically calculated flow velocity by solving the Navier–Stokes equations (in Section 3.1) using the same experimental parameters in these sets of experiments, revealing a slip velocity of ∼25 mm s−1, consistent with our previous study.33 This slip velocity can induce a very short contact time of the water stream passing a single bubble interface. The exposure time of a slippery bubble surface to water is calculated to be ∼830 μs. These short exposure times are insufficient for a gas–liquid phase equilibrium. Our findings are indicative of an additional mass transfer resistance induced by the gas dissolution at the bubble surfaces at short exposure times on a laminar flow over a slippery bubble mattress.
To further highlight our results indicative of interfacial mass transfer resistances, we present the local instantaneous convective flux distribution in Fig. 4d. The concentration profile shown in Fig. 4c and the numerical flow velocity profile (Fig. 4d, inset) are multiplied to calculate the local flux. The local flux near the bubble surfaces has low values up to y ≈ 8 μm due to the low local velocity in this region even though the dissolved oxygen concentrations are at their maximum values. The flux then increases to a peak value when 8 μm ≲ y ≲ 11 μm. Above y ≈ 11 μm, the local flux decreases with a slope mainly determined by the decrease in CO2 concentration towards the end of the diffusion layer at y ≈ 35 μm. The differences in the experimental and numerical slopes are mainly attributed to concentration profiles, as the coupling velocity profile is unique at the given settings. Here the differences in the fluxes obtained by the measurements and phase-equilibrium based simulations are more pronounced.
The O2 fluxes are determined by integration of the local flux profiles. In Fig. 5, we present the O2 fluxes obtained from the measurements, the simulations performed under phase equilibrium conditions on the bubble surfaces and the simulations incorporating an interfacial mass transfer resistance using SRT. The experimental and numerical total convective fluxes are calculated for the entire microchannel height, i.e. for 3.8 μm ≤ y ≤ H. Whereas the area under the local flux curves when 11 μm ≲ y ≲ 35 μm contributes most to the total convective flux values. It is important to note here that in this region the experimental values are not hampered by the systematic error in the measurements (y ≲ 0.1H). The experimental oxygen fluxes revealed in Fig. 5 are in a good quantitative agreement with the ones computed by considering an additional gas dissolution resistance using SRT. The flux values calculated with the assumption of a spontaneous phase-equilibrium state are higher, as in this case the interface dissolved oxygen concentration is at the maximum value. The phase equilibrium flux obtained at x = 1.05 mm for Re = 7.5 is ∼1.8 times higher than the ones obtained from the measurements and the SRT-based numerics. In Fig. 5, the oxygen fluxes obtained at Re = 10 are also presented. The deviation from the phase equilibrium mass transfer is more pronounced for a higher Reynolds number Re = 10. The phase equilibrium flux obtained at x = 1.05 mm is ∼2.2 times higher than those obtained from the measurements and the SRT-based numerics. An increase in Re from 7.5 to 10 results in a decrease in water residence time within the hold-up volume of bubble mattress from 96 ms to 48 ms, with an increase in mean velocity. Furthermore for a higher Reynolds number Re = 10, the slip velocity at the hybrid wall (y = 3 μm for θ = 35° ± 3°) has a much higher value ∼112 mm s−1, thereby a much shorter exposure time ∼180 μs of a slippery bubble surface to the water stream compared to the exposure time ∼830 μs for Re = 7.5 (Fig. 5). These findings show that these short exposure times lead to larger deviations between the equilibrium based boundary conditions (Henry's law) and the non-equilibrium based boundary conditions (SRT).
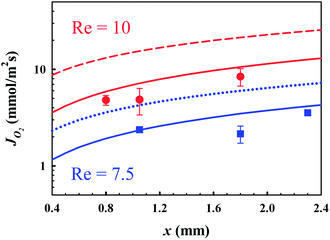 | ||
Fig. 5 O2 fluxes JO2 as functions of axial x positions and Re obtained by FLIM measurements and numerical calculations. Red filled circles ( ), the red solid line ( ), the red solid line ( ) and the red dashed line ( ) and the red dashed line ( ) indicate the experimental, the SRT-based numerical and equilibrium-based numerical results, respectively, for Re = 10 and θ = 35° ± 3°. Blue filled squares ( ) indicate the experimental, the SRT-based numerical and equilibrium-based numerical results, respectively, for Re = 10 and θ = 35° ± 3°. Blue filled squares ( ), the blue solid line ( ), the blue solid line ( ) and the blue dotted line ( ) and the blue dotted line ( ) indicate the experimental, the SRT-based numerical and equilibrium-based numerical results, respectively, for Re = 7.5 and θ = 43° ± 2°. In (a) and (b), φ = 0.38. ) indicate the experimental, the SRT-based numerical and equilibrium-based numerical results, respectively, for Re = 7.5 and θ = 43° ± 2°. In (a) and (b), φ = 0.38. | ||
We compare the experimental and gas dissolution resistance incorporated-numerical O2 fluxes obtained at x = 1.05 mm for Re = 7.5 and Re = 10. The experimental flux value for Re = 10 is ∼2.3 fold higher than that for Re = 7.5. In good agreement with the measurements, our simulations predict a ∼3 fold higher flux value for Re = 10 compared to Re = 7.5 (Fig. 5). This slight increase in the Reynolds number results in a larger decrease in exposure times due to a larger slip velocity near the bubble surface, and thereby a decrease in the dissolved oxygen concentration at the interface. Nevertheless, this enhanced hydrodynamics results in enhanced convection, and therefore an enhanced convective mass flux, as tripled in this case. Consistent with the literature findings,10,39,42,52 our findings clearly demonstrate that the hydrodynamic slippage amplifies the mass transport on hybrid substrates consisting of liquid–solid and liquid–gas interfaces.
5 Conclusions
We studied the interfacial mass transport accompanied by hydrodynamic slippage at gas–liquid interfaces in microfluidic devices. The fluidic configuration allows for the investigation of high-resolution gas absorption dynamics at curved gas–liquid interfaces. The local oxygen concentration fields resolved by FLIM measurements reveal slower gas absorption rates compared to those predicted by phase-equilibrium based simulations. The experimental results are in good agreement with the numerical results obtained from the simulations considering an additional mass transfer resistance at the gas–liquid boundary during gas dissolution. Our results indicate that the equilibrium state may not be established at short contacting times. Our findings also reveal that even at such short exposure times, the hydrodynamic slippage enhances the total mass flux driven by the amplified flow throughput on the bubble mattresses.Appendix
Additional measurements and near-bubble deviations
We verify our FLIM results by the measurements of different inlet oxygen concentrations which were prescribed to certain values prior to microfluidic mass transfer experiments by the aid of an external oxygen sensor during the bulk solution preparation. Fig. 6a and b show the dissolved oxygen concentration profiles across the microchannel height, with respect to the y direction normal to the bubble surfaces for varying inlet concentrations of 0.5 and 0.2 mol m−3. The experimental results are compared with numerical results obtained for phase-equilibrium based and SRT-based gas absorption.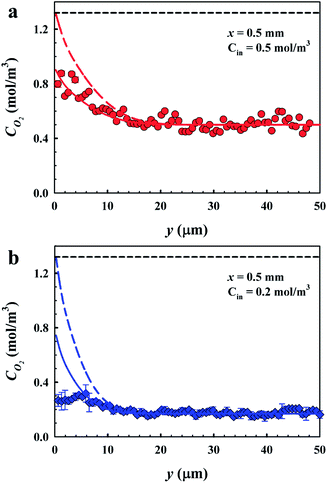 | ||
| Fig. 6 Experimental and numerical local dissolved oxygen profiles. (a) When the inlet oxygen concentration is 0.5 mol m−3. Here Re = 2.2. (b) When the inlet oxygen concentration is 0.2 mol m−3. Here Re = 4. In (a and b), symbols represent the experimental results obtained by FLIM, the colored solid lines represent the numerical results solved with SRT, and the colored dashed lines represent the numerical results solved using Henry's law. Here H = 50 μm, θ = 2° ± 1° and φ = 0.54. The black dashed lines depict the equilibrium saturation value of oxygen in water. | ||
The bubble interface position is y ∼ 0.3 μm as shown in Fig. 6a, as the bubbles are nearly flat (θ = 2° ± 1°). At the bubble interface (y ∼ 0.3 μm), the oxygen concentration is measured to be ∼0.9 mol m−3, consistent with the SRT boundary condition numerical results. Whereas the interface O2 concentration predicted by phase-equilibrium based simulations converges to a saturation value of 1.3 mol m−3. The thickness of the diffusion boundary layer obtained by FLIM measurements extends to ∼18 μm, consistent with the numerical results. Beyond the thickness of the diffusion boundary layer, the dissolved oxygen concentration drops to an inlet bulk concentration of 0.5 mol m−3. Similarly, the experimental oxygen concentration profile shown in Fig. 6b reaches an inlet bulk concentration of 0.2 mol m−3 beyond the diffusion boundary layer thickness at y ∼ 14 μm. In Fig. 6b, the measured O2 profile is in good agreement with SRT based simulations when y ≳ 4 μm. However there is a discrepancy between the measurements and both numerical results in the proximity of the bubble interfaces, as shown in Fig. 4. The standard errors close to the bubble surface are larger compared to those of the remainder of the microchannel. The discrepancy in the measured and calculated values when y ≲ 4 μm is still larger than the standard error margins.
The near-bubble deviations can be related to the systematic error in our FLIM measurements close to the gas–liquid interfaces. In the FLIM measurements, the normalized fluorescence lifetime signal decreases from 1 (oxygen-free state) to ∼0.3 (oxygen-saturated state), providing a signal level of ∼0.7 in the entire oxygen concentration range. We measure the noise level to be ∼0.1, hence a signal to noise ratio of ∼7. However we observed reduced signal-to-noise ratios near microchannel walls and bubble interfaces. Such deviations near gas–liquid interfaces have also been reported in previous reports53–56 and are attributed to optical distortions and reflection effects near interfaces. Besides, we observed differences in the range of ≈7–15% between the lifetime data calculated by the phase shift and that of the amplitude demodulation of the emitted signal near interfaces. Differences in the phase shift τp and demodulation τm lifetimes have been reported to be indicative of complex quenching mechanisms and are observed for long lifetime probes as Ru-based complexes in previous studies.57
Acknowledgements
Grants from The Netherlands Organization for Scientific Research-ACTS for a PhD fellowship (to E.K.) (Process on a Chip Project 053.65.007) and from European Research Council (an ERC starting grant 307342-TRAM to R.G.H.L.) are gratefully acknowledged.References
- Perry's Chemical Engineers' Handbook, ed. D. W. Green, McGraw Hill, 6th edn, 1984 Search PubMed.
- M. Adler, M. Erickstad, E. Gutierrez and A. Groisman, Lab Chip, 2012, 12, 4835–4847 RSC.
- B. Ungerböck, A. Pohar, T. Mayr and I. Plazl, Microfluid. Nanofluid., 2013, 14, 565–574 CrossRef.
- Transport Processes and Separation Process Principles, ed. C. J. Geankoplis, Prentice Hall, 4th edn, 2003 Search PubMed.
- E. S. Gaddis, Chem. Eng. Process., 1999, 38, 503–510 CrossRef CAS.
- W. Henry, Philos. Trans. R. Soc. London, 1803, 93, 29–42 CrossRef , 274–276.
- S. Karoor and K. K. Sirkar, Ind. Eng. Chem. Res., 1993, 32, 674–684 CrossRef CAS.
- J. de Jong, P. W. Verheijden, R. G. H. Lammertink and M. Wessling, Anal. Chem., 2008, 80, 3190–3197 CrossRef CAS PubMed.
- J. de Jong, R. G. H. Lammertink and M. Wessling, Lab Chip, 2006, 6, 1125–1139 RSC.
- E. Karatay and R. G. H. Lammertink, Lab Chip, 2012, 12, 2922–2929 RSC.
- Membrane Handbook, ed. W. S. W. Ho and K. K. Sirkar, Chapman and Hall, 1992, vol. 1, ch. 46 Search PubMed.
- M. Mavroudi, S. Kaldis and G. Sakellaropoulos, J. Membr. Sci., 2006, 272, 103–115 CrossRef CAS PubMed.
- H. Y. Zhang, R. Wang, D. T. Liang and J. H. Tay, J. Membr. Sci., 2008, 308, 162–170 CrossRef CAS PubMed.
- V. Dindore, D. Brilman and G. Versteeg, J. Membr. Sci., 2005, 251, 209–222 CrossRef CAS PubMed.
- H. Kreulen, C. Smolders, G. Versteeg and W. van Swaaij, J. Membr. Sci., 1993, 78, 197–216 CrossRef CAS.
- E. van Elk, P. Borman, J. Kuipers and G. Versteeg, Chem. Eng. J., 2000, 76, 223–237 CrossRef CAS.
- E. van Elk, M. Knaap and G. Versteeg, Chem. Eng. Res. Des., 2007, 85, 516–524 CrossRef CAS.
- W. G. Whitman, Chem. Metall. Eng., 1923, 29, 146–148 CAS.
- R. Higbie, Trans. Am. Inst. Chem. Eng., 1935, 35, 36–60 Search PubMed.
- H. L. Toor and J. M. Marchello, AIChE J., 1958, 4, 97–101 CrossRef CAS.
- P. V. Danckwerts, Ind. Eng. Chem., 1951, 43, 1460–1467 CrossRef CAS.
- T. Huthwelker and T. Peter, J. Chem. Phys., 1996, 105, 1661–1667 CrossRef CAS.
- L. Monchick and R. Blackmore, J. Aerosol Sci., 1988, 19, 273–286 CrossRef CAS.
- C. A. Ward, R. D. Findlay and M. Rizk, J. Chem. Phys., 1982, 76, 5599–5605 CrossRef CAS.
- C. A. Ward, M. Rizk and A. S. Tucker, J. Chem. Phys., 1982, 76, 5606–5614 CrossRef CAS.
- C. A. Ward, J. Chem. Phys., 1977, 67, 229–235 CrossRef CAS.
- P. Rahimi and C. Ward, Int. J. Heat Mass Transfer, 2004, 47, 877–886 CrossRef CAS PubMed.
- R. Holyst, et al. , Rep. Prog. Phys., 2013, 76, 034601 CrossRef CAS PubMed.
- P. Rahimi and C. Ward, Int. J. Thermodyn., 2005, 8, 1–14 Search PubMed.
- V. K. Badam, V. Kumar, F. Durst and K. Danov, Exp. Therm. Fluid Sci., 2007, 32, 276–292 CrossRef CAS PubMed.
- J. A. W. Elliott and C. A. Ward, J. Chem. Phys., 1997, 106, 5667–5676 CrossRef CAS.
- P. Tsai, et al. , Phys. Fluids, 2009, 21, 112002 CrossRef.
- E. Karatay, et al. , Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8422–8426 CrossRef CAS PubMed.
- A. Steinberger, C. Cottin-Bizonne, P. Kleimann and E. Charlaix, Nat. Mater., 2007, 6, 665–668 CrossRef CAS PubMed.
- D. Byun, J. Kim, H. S. Ko and H. C. Park, Phys. Fluids, 2008, 20, 113601 CrossRef.
- C. J. Teo and B. C. Khoo, Microfluid. Nanofluid., 2010, 9, 499–511 CrossRef.
- C. Ng and C. Y. Wang, Fluid Dyn. Res., 2011, 43, 065504 CrossRef.
- S. Schmieschek, A. V. Belyaev, J. Harting and O. I. Vinogradova, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 85, 016324 CrossRef.
- L. Bocquet and J.-L. Barrat, Soft Matter, 2007, 3, 685–693 RSC.
- J. Hyväluoma and J. Harting, Phys. Rev. Lett., 2008, 100, 246001 CrossRef.
- L. Bocquet and E. Lauga, Nat. Mater., 2011, 10, 334–337 CrossRef CAS PubMed.
- A. Ajdari and L. Bocquet, Phys. Rev. Lett., 2006, 96, 186102 CrossRef.
- R. Arayanarakool, L. Shui, A. van den Berg and J. C. T. Eijkel, Lab Chip, 2011, 11, 4260–4266 RSC.
- H. C. Gerritsen, R. Sanders, A. Draaijer, C. Ince and Y. K. Levine, J. Fluoresc., 1997, 7, 11–15 CrossRef CAS.
- P. Abbyad, et al. , Lab Chip, 2010, 10, 2505–2512 RSC.
- M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641–2684 CrossRef CAS PubMed.
- K. J. Morris, et al. , Anal. Chem., 2007, 79, 9310–9314 CrossRef CAS PubMed.
- M. Polinkovsky, E. Gutierrez, A. Levchenko and A. Groisman, Lab Chip, 2009, 9, 1073–1084 RSC.
- D. Sud, W. Zhong, D. G. Beer and M.-A. Mycek, Opt. Express, 2006, 14, 4412–4426 CrossRef CAS.
- G. Mehta, et al. , Biomed. Microdevices, 2007, 9, 123–134 CrossRef CAS PubMed.
- J. Chen, H. D. Kim and K. C. Kim, Microfluid. Nanofluid., 2013, 14, 541–550 CrossRef CAS.
- H. M. Yeh and T. W. Cheng, J. Membr. Sci., 1999, 154, 41–51 CrossRef CAS.
- J. W. Walker and W. L. Peirson, Exp. Fluids, 2008, 44, 249–259 CrossRef CAS PubMed.
- J. Philip, T. Woodrow and S. R. Duke, Ind. Eng. Chem. Res., 2001, 40, 1985–1995 CrossRef.
- S. Roy and S. R. Duke, Rev. Sci. Instrum., 2000, 71, 3494–3501 CrossRef CAS.
- G. H. Jirka, H. Herlina and A. Niepelt, Exp. Fluids, 2010, 49, 319–327 CrossRef CAS PubMed.
- H. Choi, et al. , Opt. Express, 2012, 20, 26219–26235 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2013 |