
A new double-reference correction scheme for accurate and efficient computation of NMR chemical shieldings†

Abstract
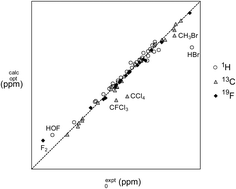
Using an extensive set of benchmark geometric data and absolute NMR chemical shieldings for gas phase molecules compiled from the literature, we introduce and test a novel shift-and-scale correction scheme for bringing computed values into closer agreement with experiment. Our approach requires only one additional reference calculation relative to existing methods for computing chemical shifts, and all reference species are small molecules whose absolute nuclear shieldings can be easily obtained, both computationally and experimentally. We demonstrate that our approach is capable of correcting for errors due to choice of electronic structure method and basis set, vibrational averaging effects, and scalar relativistic effects, but cannot account for the influence that heavy atoms have on the chemical shieldings of neighbouring light atoms, via spin–orbit coupling. We particularly recommend using our approach in conjunction with nuclear shieldings computed at DFT optimized geometries, employing hybrid functionals with moderate-to-high quality atomic orbital basis sets, e.g. pc-2.
- This article is part of the themed collections: Showcasing Physical Chemistry research in Australia and New Zealand, Benchmark Experiments for Numerical Quantum Chemistry and 2022 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

