
A current overview of the oxidative desulfurization of fuels utilizing heat and solar light: from materials design to catalysis for clean energy
 ab
and
Wee-Jun
Ong
*abc
ab
and
Wee-Jun
Ong
*abc
Abstract
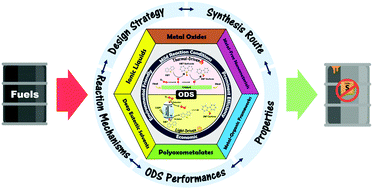
The ceaseless increase of pollution cases due to the tremendous consumption of fossil fuels has steered the world towards an environmental crisis and necessitated urgency to curtail noxious sulfur oxide emissions. Since the world is moving toward green chemistry, a fuel desulfurization process driven by clean technology is of paramount significance in the field of environmental remediation. Among the novel desulfurization techniques, the oxidative desulfurization (ODS) process has been intensively studied and is highlighted as the rising star to effectuate sulfur-free fuels due to its mild reaction conditions and remarkable desulfurization performances in the past decade. This critical review emphasizes the latest advances in thermal catalytic ODS and photocatalytic ODS related to the design and synthesis routes of myriad materials. This encompasses the engineering of metal oxides, ionic liquids, deep eutectic solvents, polyoxometalates, metal–organic frameworks, metal-free materials and their hybrids in the customization of advantageous properties in terms of morphology, topography, composition and electronic states. The essential connection between catalyst characteristics and performances in ODS will be critically discussed along with corresponding reaction mechanisms to provide thorough insight for shaping future research directions. The impacts of oxidant type, solvent type, temperature and other pivotal factors on the effectiveness of ODS are outlined. Finally, a summary of confronted challenges and future outlooks in the journey to ODS application is presented.
- This article is part of the themed collections: Nanoscale Horizons and Nanoscale: Nanomaterials for Energy and Horizons Community Board Collection: Solar Energy Conversion
 Please wait while we load your content...
Please wait while we load your content...

