
G4 accuracy at DFT cost: unlocking accurate redox potentials for organic molecules using systematic error cancellation†
 a
and
Krishnan
Raghavachari
*a
a
and
Krishnan
Raghavachari
*a
Abstract
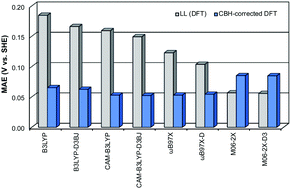
The redox potential is a powerful thermodynamic and kinetic tool used to predict numerous chemical and biochemical mechanisms. However, despite the improving predictive power of density functional theory (DFT), chemically accurate theoretical redox potentials are often difficult to achieve with DFT. For example, calculated redox potentials are sensitive to density functional choice and often fall short of the desired accuracy. Thus, ranges of errors for computed redox potentials between different density functionals can become quite large. The current study presents a cost-effective protocol that utilizes effective error cancellation schemes in order to accurately predict the redox potentials of a wide range of organic molecules. This computational protocol, called CBH-Redox, is an extension of the connectivity-based hierarchy (CBH) method, and produces thermochemical data with near-G4 accuracy. Herein, we test the CBH-Redox protocol against both experimental and G4 reference values and compare these results to DFT alone. Considering 46 C, O, N, F, Cl, and S atom-containing molecules, when using the CBH-Redox correction scheme, the MAEs for all eight density functionals tested are within the 0.09 V target accuracy versus both experiment and G4. Moreover, CBH-Redox achieves an impressive accuracy, with a MAE of 0.05 V or below when compared to G4 for six of the eight density functionals tested. In addition, when the CBH correction is applied, the error range across all functionals tested decreases from 0.12 V to about 0.05 V versus G4, and from 0.13 V to 0.04 V versus experiment.
- This article is part of the themed collection: 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

