
Understanding photophysical properties of iridium complexes with N-(5-phenyl-1,3,4-oxadiazol-2-yl)-diphenylphosphinic amide as the ancillary ligand†
 *a
Guochun
Yang
*b
*a
Guochun
Yang
*b
Abstract
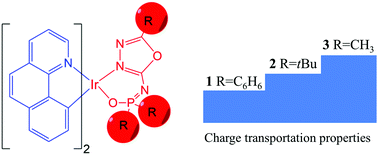
Understanding the physical nature behind experimental phenomena is rather significant to further optimize the performance of a material. Now, density functional theory (DFT) has become a well-accepted tool for unveiling the origin of physical/chemical properties of materials and designing new materials with desirable properties. Recently, two novel POXD-based (N-(5-phenyl-1,3,4-oxadiazol-2-yl)-diphenylphosphinic amide) iridium(III) complexes 1 and 4 with different cyclometalated ligands showed significant performance differences. Herein, we employ DFT calculations to investigate the electronic structures, absorption and emission spectra, and charge transportation properties of iridium(III) complexes 1 and 4. In comparison with 4, the good performance of 1 can be attributed to its favorable charge transport properties. Based on complex 1, two new iridium complexes (2 and 3) were theoretically designed by substituting phenyl rings with a tert-butyl group (-t-Bu) and methyl group (–CH3), respectively. The results clearly indicated that the -t-Bu and –CH3 groups enhance the hole and electron injection abilities and improve the charge balance. On the other hand, the designed complexes 2 and 3 also show a blue-shift in emission spectra with respect to complex 1. As a result, complexes 2 and 3 are expected to be promising phosphorescence emitters with good device performance.
 Please wait while we load your content...
Please wait while we load your content...

