
Cooperative nature of the sulfur centered hydrogen bond: investigation of (H2S)n (n = 2–4) clusters using an affordable yet accurate level of theory†

Abstract
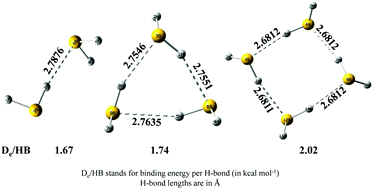
Existing studies have shown that appreciably high level quantum chemical calculations are required to reproduce experimental energetic and geometric features of a H2S dimer. This condition severely restricts any practical possibility of obtaining reliable results for higher order H2S clusters. We have shown here that the binding energies calculated at the CCSD(T)/CBS level with counterpoise corrected geometries calculated at the MP2/aug-cc-pV(Q+d)Z level of theory excellently match with the experimental results for the H2S dimer. Subsequently, the above mentioned levels of theory were used for trimers and tetramers. (H2S)n (n = 2–4) clusters were found to show cooperative strengthening of S–H⋯S hydrogen bonds, which is clearly evident from the evolution of binding energies and hydrogen bond lengths, with increasing cluster size. Localized molecular orbital energy decomposition analyses have been carried out to understand how the contributions of various energy components modulate with the size of the clusters and what are their relative contributions towards the overall stabilization of the clusters. Natural bond orbital and atoms in molecules analyses were also carried out in order to look into the evolution of the electronic charge transfer and electron density topology with cluster size.
 Please wait while we load your content...
Please wait while we load your content...

