
Unexpected reversal of stability in strained systems containing one-electron bonds†
 abc
Wania
Wolff,
*d
Heidy M.
Quitián-Lara,
e
Heloisa M.
Boechat-Roberty,
e
Marco Antonio Chaer
Nascimento
*a
abc
Wania
Wolff,
*d
Heidy M.
Quitián-Lara,
e
Heloisa M.
Boechat-Roberty,
e
Marco Antonio Chaer
Nascimento
*a
Abstract
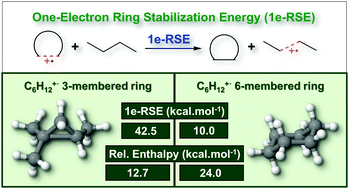
Ring strain energy is a very well documented feature of neutral cycloalkanes, and influences their structural, thermochemical and reactivity properties. In this work, we apply density functional theory and high-level coupled cluster calculations to describe the geometry and relative stability of C6H12+˙ radical cations, whose cyclic isomers are prototypes of singly-charged cycloalkanes. Molecular ions with the mentioned stoichiometry were produced via electron impact experiments using a gaseous cyclohexane sample (20–2000 eV). From our calculations, in addition to structures that resemble linear and branched alkenes as well as distinct conformers of cyclohexane, we have found low-lying species containing three-, four- and five-membered rings with the presence of an elongated C–C bond. Remarkably, the stability trend of these ring-bearing radical cations is anomalous, and the three-membered species are up to 11.3 kcal mol−1 more stable than the six-membered chair structure. Generalized Valence Bond calculations and the Spin Coupled theory with N electrons and M orbitals were used in conjunction with the Generalized Product Function Energy Partitioning (GPF-EP) method and Interference Energy Analysis (IEA) to describe the chemical bonding in such moieties. Our results confirm that these elongated C–C motifs are one-electron sigma bonds. Our calculations also reveal the effects that drive thermochemical preference of strained systems over their strained-free isomers, and the origin of the unusual stability trend observed for cycloalkane radical cations.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

