
Band alignment in quantum wells from automatically tuned DFT+U
 †*ab
†*ab
Abstract
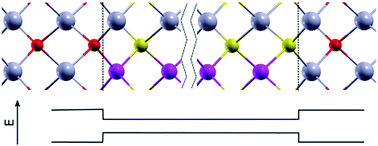
Band alignment between two materials is of fundamental importance for a multitude of applications. However, density functional theory (DFT) either underestimates the bandgap – as is the case with the local density approximation (LDA) or generalized gradient approximation (GGA) – or is highly computationally demanding, as is the case with hybrid-functional methods. The latter can become prohibitive in electronic-structure calculations of supercells which describe quantum wells. We propose to apply the DFT+U method, with U for each atomic shell being treated as set of tuning parameters, to automatically fit the bulk bandgap and the lattice constant, and then use the thus obtained U parameters in large supercell calculations to determine the band alignment. We apply this procedure to InP/In0.5Ga0.5As, In0.5Ga0.5As/In0.5Al0.5As and InP/In0.5Al0.5As quantum wells, and obtain good agreement with experimental results. Although this procedure requires some experimental input, it provides both meaningful valence and conduction band offsets while, crucially, lattice relaxation is taken into account. The computational cost of this procedure is comparable to that of LDA. We believe that this is a practical procedure that can be useful for providing accurate estimates of band alignments between more complicated alloys.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

