
Assessing AMBER force fields for protein folding in an implicit solvent†
 *a
and
*a
and
Abstract
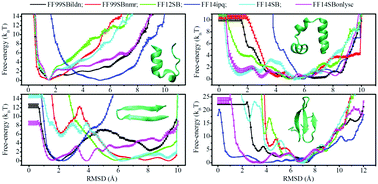
Molecular dynamics (MD) simulation implemented with a state-of-the-art protein force field and implicit solvent model is an attractive approach to investigate protein folding, one of the most perplexing problems in molecular biology. But how well can force fields developed independently of implicit solvent models work together in reproducing diverse protein native structures and measuring the corresponding folding thermodynamics is not always clear. In this work, we performed enhanced sampling MD simulations to assess the ability of six AMBER force fields (FF99SBildn, FF99SBnmr, FF12SB, FF14ipq, FF14SB, and FF14SBonlysc) as coupled with a recently improved pair-wise GB-Neck2 model in modeling the folding of two helical and two β-sheet peptides. Whilst most of the tested force fields can yield roughly similar features for equilibrium conformational ensembles and detailed folding free-energy profiles for short α-helical TC10b in an implicit solvent, the measured counterparts are significantly discrepant in the cases of larger or β-structured peptides (HP35, 1E0Q, and GTT). Additionally, the calculated folding/unfolding thermodynamic quantities can only partially match the experimental data. Although a combination of the force fields and GB-Neck2 implicit model able to describe all aspects of the folding transitions towards the native structures of all the considered peptides was not identified, we found that FF14SBonlysc coupled with the GB-Neck2 model seems to be a reasonably balanced combination to predict peptide folding preferences.
 Please wait while we load your content...
Please wait while we load your content...

