
Influence of molecular weight between crosslinks on the mechanical properties of polymers formed via ring-opening metathesis†
 a
Daniel B.
Knorr, Jr.
*a
and
a
Daniel B.
Knorr, Jr.
*a
and
Abstract
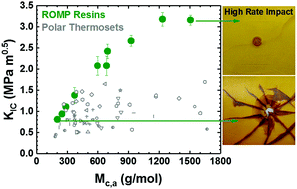
The apparent molecular weight between crosslinks (Mc,a) in a polymer network plays a fundamental role in the network mechanical response. We systematically varied Mc,a independent of strong noncovalent bonding by using ring-opening metathesis polymerization (ROMP) to co-polymerize dicyclopentadiene (DCPD) with a chain extender that increases Mc,a or a di-functional crosslinker that decreases Mc,a. We compared the ROMP series quasi-static modulus (E), tensile yield stress (σy), and fracture toughness (KIC and GIC) in the glassy regime with literature data for more polar thermosets. ROMP resins showed high KIC (>1.5 MPa m0.5), high GIC (>1000 J m−2), and 4–5 times higher high rate impact resistance than typical polar thermosets with similar Tg values (100 °C to 178 °C). The overall E values were lower for ROMP systems. The σy dependence on Mc,a and T–Tg for ROMP resins was qualitatively similar to more polar thermosets, but the overall σy values were lower. In contrast to more polar thermosets, the KIC and GIC values of the ROMP resins showed strong Mc,a and T–Tg dependence. High rate impact (∼104–105 s−1) trends were similar to the KIC and GIC behavior, but were also correlated to σy. Overall, a ductile failure mode was observed for quasi-static and high rate results for a linear ROMP polymer (Mc,a = 1506 g mol−1 due to chain entanglement), and this gradually transitioned to a fully brittle failure mode for highly crosslinked ROMP polymers (Mc,a ≤ 270 g mol−1). Molecular dynamics (MD) simulations showed that low Mc,a ROMP resins were more likely to form molecular scale nanovoids. The higher chain stiffness in low Mc,a ROMP resins inhibited stress relaxation in the vicinity of these nanovoids, which correlated with brittle mechanical responses. Overall, these differences in mechanical properties were attributed to the weak non-covalent interactions in ROMP resins.
 Please wait while we load your content...
Please wait while we load your content...

