
Ab initio study of medium sized boron-doped silicon clusters SinBm, n = 11–13, m = 1–3†

Abstract
The ground state and energetically low structures of neutral SinBm clusters, of medium size with n = 11–13, m = 1–3, are identified, presented and rationalized. Structures of the nanoclusters are predicted using density functional theory (DFT) and employing the HSE06 range-separated hybrid exchange–correlation functional. For these systems the functional is shown to offer systematic performance when benchmarked against high accuracy coupled-cluster CCSD(T) and compared to well known functionals used in the literature. Discrepancies for small size systems present in the literature are addressed and resolved. The structural evolution patterns of the clusters are discussed and common structural features (substructures) are identified. Cluster geometries are extensively searched via a particle swarm optimization algorithm alongside more traditional methodologies. In addition to the binding energies (that include zero-point energy corrections) of the structures, the optical gaps and UV/visible absorption spectra are reported, employing the CAM-B3LYP functional that was benchmarked against the high level EOM-CCSD level of theory. The computed infrared spectra are provided and discussed in length with respect to structural details. Their effectiveness as charge transfer units is examined. Optical gaps range between 1.4–2.5 eV, and are adjustable through the boron and silicon content of the clusters, which, along with the increased structural stability, offers promise for applications in optoelectronics.
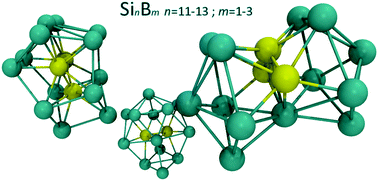
 Please wait while we load your content...
Please wait while we load your content...

