
Time-resolved photoelectron spectroscopy of IR-driven electron dynamics in a charge transfer model system
 *a
and
Stefanie
Gräfe
*b
*a
and
Stefanie
Gräfe
*b
Abstract
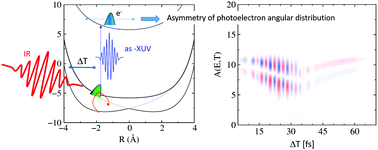
If the adiabatic approximation is valid, electrons smoothly adapt to molecular geometry changes. In contrast, as a characteristic of diabatic dynamics, the electron density does not follow the nuclear motion. Recently, we have shown that the asymmetry in time-resolved photoelectron spectra serves as a tool to distinguish between these dynamics [Falge et al., J. Phys. Chem. Lett., 2012, 3, 2617]. Here, we investigate the influence of an additional, moderately intense infrared (IR) laser field, as often applied in attosecond time-resolved experiments, on such asymmetries. This is done using a simple model for coupled electronic-nuclear motion. We calculate time-resolved photoelectron spectra and their asymmetries and demonstrate that the spectra directly map the bound electron–nuclear dynamics. From the asymmetries, we can trace the IR field-induced population transfer and both the field-driven and intrinsic (non-)adiabatic dynamics. This holds true when considering superposition states accompanied by electronic coherences. The latter are observable in the asymmetries for sufficiently short XUV pulses to coherently probe the coupled states. It is thus documented that the asymmetry is a measure for phases in bound electron wave packets and non-adiabatic dynamics.
- This article is part of the themed collection: XUV/X-ray light and fast ions for ultrafast chemistry
 Please wait while we load your content...
Please wait while we load your content...

