
Towards the prediction of the transport properties of cluster-based molybdenum chalcogenides†

Abstract
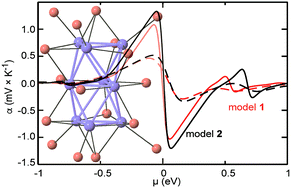
Molybdenum cluster chemistry is one of the richest series of cluster-based materials with nuclearities of up to 40. Among them, molybdenum chalcogenides are promising materials for high-temperature thermoelectric applications due to their intrinsic, extremely low thermal conductivities. In this paper, we studied the electronic transport properties of three selenides based on octahedral Mo6 and bioctahedral Mo9 motifs using band structure calculations and a semi-classical approach. The electronic structure of these materials is governed by the cluster units. Unlike the electronic conductivity, the computed thermopower hardly depends on the structural details suggesting that such a computational approach may be useful to identify new interesting candidates for thermoelectric applications among cluster-based materials.
 Please wait while we load your content...
Please wait while we load your content...

