
Understanding the pH-dependent adsorption of ionizable compounds on graphene oxide using molecular dynamics simulations†
 *ab
and
Baoshan
Xing
*c
*ab
and
Baoshan
Xing
*c
Abstract
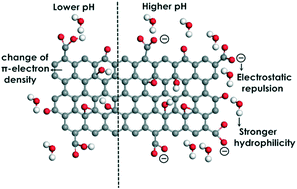
Bisphenol A was used as a representative of ionizable organic compounds (IOCs) to investigate their adsorption on graphene oxide (GO). To explore the pH-dependent adsorption of IOCs on GO, analysis of electrostatic interaction energy, hydrophobicity, and π-electron densities of benzene rings under different pH conditions was carried out using molecular dynamics (MD) simulations and density functional theory (DFT) calculations. Six H-bond configurations were witnessed during the adsorption process: (1) intramolecular H-bonds between functional groups of GO; (2) GO⋯water H-bonds; (3) GO⋯water⋯GO H-bonds; (4) IOC⋯GO H-bonds; (5) IOC⋯water⋯GO H-bonds; and (6) IOC⋯water H-bonds. MD results indicated that more H-bonds appeared between GO (or IOC) and water than between IOC and GO, indicating that the water-mediated H-bond acted as a steering force in the adsorption process. Upon adsorption, the configurations of IOCs and GO were changed, and the hydrophilicity was decreased, which may in turn affect their fate and toxicity in the environment. The change of π-electron density of several common IOCs was provided, which may help to understand the pH-dependent behaviour of IOCs in future studies.
 Please wait while we load your content...
Please wait while we load your content...

