
Enhancement in catalytic proton reduction by an internal base in a diiron pentacarbonyl complex: its synthesis, characterisation, inter-conversion and electrochemical investigation†
 *b
*b
Abstract
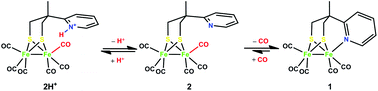
The reaction of a tripodal ligand (H2L) with a {S2N} donor-set with tri-iron dodecacarbonyl in toluene leads to the isolation of a diiron pentacarbonyl complex 1 as a model for the sub-site of the [FeFe]-hydrogenase. Protonation of this complex under CO (1 atm.) forms quantitatively the hexacarbonyl complex 2H+ with a pendant pyridinium group. Infrared spectroscopic investigations indicate that its pendant pyridinium group dissociates to give hexacarbonyl complex 2 which forms subsequently the pentacarbonyl complex 1. The electrochemistry of these complexes has been investigated. Complex 2H+ exhibits electrocatalysis on proton reduction at a potential more positive by over 200 mV compared to that for other neutral diiron hexacarbonyl complexes. This catalysis is enhanced under a CO atmosphere by freeing the bound base group which acts as a proton relay in the catalysis.
 Please wait while we load your content...
Please wait while we load your content...

