
First principles prediction of CH4 reactivities with Co3O4 nanocatalysts of different morphologies†
 *ac
and
*ac
and
Abstract
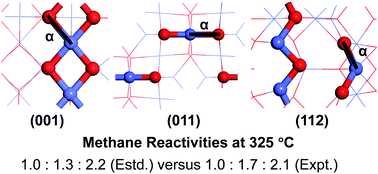
Co3O4 nanocatalysts have been experimentally shown to have excellent performance in catalyzing CH4 combustion. These nanocatalysts of different morphologies, such as nanoparticle/nanocube, nanorod/nanobelt, and nanoplate/nanosheet, were previously synthesized and characterized to mainly expose the (001), (011), and (112) surfaces, respectively, with distinct reactivities. In this study, rigorous first principles calculations were performed to investigate CH4 reactivities of the above Co3O4 surfaces of different terminations. CH4 dissociation was predicted to occur at the Co–O pair site on these surfaces. For each surface, the most reactive Co–O pair site was identified based on calculated energy barriers of the different active sites, which should contribute most significantly to the reactivity of that surface. The lowest energy barriers for the (001), (011), and (112) surfaces were predicted to be 0.96, 0.90, and 0.79 eV, respectively, suggesting CH4 reactivity to increase in that order for the different Co3O4 surfaces, consistent with the trend found experimentally for Co3O4 nanocatalysts of different morphologies. Direct comparison between the estimated and experimental CH4 reaction rates per gram of the nanocatalysts at 325 °C further indicate that their relative ratios were well reproduced by considering three main factors: the effective energy barrier for CH4 dissociation, the surface area of the nanocatalyst, and the number of independent active sites per unit surface area. The important influence of surface area on CH4 reactivity is also demonstrated by the significant difference in the reactivities of the nanocatalysts when exposing the same facet but with distinct surface areas.
 Please wait while we load your content...
Please wait while we load your content...

