
Theoretical insights into the effect of terrace width and step edge coverage on CO adsorption and dissociation over stepped Ni surfaces†
 *a
*a
Abstract
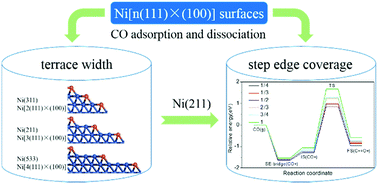
Vicinal surfaces of Ni are model catalysts of general interest and great importance in computational catalysis. Here we report a comprehensive study conducted with density functional theory on Ni[n(111) × (100)] (n = 2, 3 and 4) surfaces to explore the effect of terrace width and step edge coverage on CO adsorption and dissociation, a probe reaction relevant to many industrial processes. The coordination numbers (CN), the generalized coordination numbers 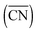 and the d band partial density of states (d-PDOS) of Ni are identified as descriptors to faithfully reflect the difference of the step edge region for Ni[n(111) × (100)]. Based on analysis of the energy diagrams for CO activation and dissociation as well as the structural features of the Ni(311), Ni(211) and Ni(533) surfaces, Ni(211) (n = 3) is proposed as a model of adequate representativeness for Ni[n(111) × (100)] (n ≥ 3) surface groups in investigating small molecule activation over such stepped structures. Further, a series of Ni(211) surfaces with the step edge coverage ranging from 1/4 to 1 monolayer (ML) were utilized to assess their effect on CO activation. The results show that CO adsorption is not sensitive to the step edge coverage, which could readily approach 1 ML under a CO-rich atmosphere. In contrast, CO dissociation manifests strong coverage dependence when the coverage exceeds 1/2 ML, indicating that significant adsorbate–adsorbate interactions emerge. These results are conducive to theoretical studies of metal-catalyzed surface processes where the defects play a vital role.
and the d band partial density of states (d-PDOS) of Ni are identified as descriptors to faithfully reflect the difference of the step edge region for Ni[n(111) × (100)]. Based on analysis of the energy diagrams for CO activation and dissociation as well as the structural features of the Ni(311), Ni(211) and Ni(533) surfaces, Ni(211) (n = 3) is proposed as a model of adequate representativeness for Ni[n(111) × (100)] (n ≥ 3) surface groups in investigating small molecule activation over such stepped structures. Further, a series of Ni(211) surfaces with the step edge coverage ranging from 1/4 to 1 monolayer (ML) were utilized to assess their effect on CO activation. The results show that CO adsorption is not sensitive to the step edge coverage, which could readily approach 1 ML under a CO-rich atmosphere. In contrast, CO dissociation manifests strong coverage dependence when the coverage exceeds 1/2 ML, indicating that significant adsorbate–adsorbate interactions emerge. These results are conducive to theoretical studies of metal-catalyzed surface processes where the defects play a vital role.
 Please wait while we load your content...
Please wait while we load your content...

