
Dehydro-oxazole, thiazole and imidazole radicals: insights into the electronic structure, stability and reactivity aspects†
 a
and
a
and
Abstract
DFT (U)B3LYP/cc-pVTZ and (U)M06-2X/cc-pVTZ and multireference CASSCF/cc-pVTZ level of theories have been used to investigate the electronic structure of isomeric dehydrooxazole (1b–d, 3 isomers), dehydrothiazole (2b–d, 3 isomers) and dehydroimidazole (3a–d, 4 isomers) radicals. The ground state electronic structure of each radical isomer has been confirmed by predicting their doublet excited state structure and calculating the adiabatic energy difference. The stability order of the individual isomeric radicals has been estimated through the comparison of absolute energies. A hypothetical isodesmic reaction has been utilized to calculate radical stabilization energies (RSEs). The results show N-dehydro imidazole 3a as the thermodynamically most stable radical. In order to understand the structural and stability aspects of the radicals and interactions between the radical electron and the electron lone pairs, we have analysed spin densities and hybridisation changes and also performed MCSCF calculations. The reason for the higher stability of 3a has been attributed to the attainment of a π-character and subsequent delocalization, whereas, all other carbon centred radicals are found to be localized σ-radicals. Furthermore, the kinetic stability of the radicals has been investigated through unimolecular decomposition channels. All the studies showed a weak to strong coupling between the N3 and the radical centre depending on the location of the radical centre. NBO analysis suggests that through space coupling between N3 and σ* of the radical centre leads to a stabilising effect when the radical centre is adjacent to N3, whereas such interactions are absent when the radical centre is away from it. However, only a weak coupling is observed between the radical centre and X1 (O/S/NH). Particularly the interaction strength has the following trend: S < O < N–H. Indeed S, O and N–H show a stabilising effect through bond interactions with the antibonding orbitals of the alternate bonds on either side.
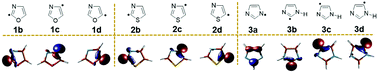
 Please wait while we load your content...
Please wait while we load your content...

