
Nature of S2Se2 σ(4c–6e) at naphthalene 1,8-positions and models, elucidated by QTAIM dual functional analysis†
 *a
*a
Abstract
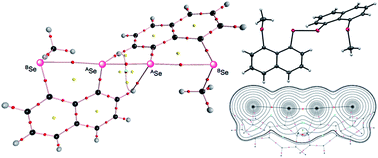
The nature of extended hypervalent interactions of the BE–*–AE–*–AE–*–BE type is elucidated for 1-(8-MeBEC10H6)AE–AE(C10H6BEMe-8′)-1′, (1 (AE, BE) = (S, S), 2 (S, Se), 3 (Se, S) and 4 (Se, Se)) and models A–D, BR2BE⋯(AR)AE–AE(AR)⋯BEBR2 (AR, BR = H and Me). QTAIM dual functional analysis, which we proposed recently, is applied to the analysis. Total electron energy densities Hb(rc) are plotted versus Hb(rc) − Vb(rc)/2 for the interactions at bond critical points (BCPs; *), where Vb(rc) show potential energy densities at BCPs. Data for the perturbed structures around the fully optimized structures are employed for the plots, in addition to those of the fully optimized ones. While the data for the fully optimized structures are analysed by the polar coordinate (R, θ) representation, those containing the perturbed structures are by (θp, κp): θp corresponds to the tangent line for the plot and κp is the curvature. While (R, θ) show the static nature, (θp, κp) represent the dynamic nature of interactions. All AE–*–AE interactions in 1–4 and models A–D are classified by the shard shell interactions and have the character of a weak covalent nature. The AE–*–BE interactions in 1–4 are all classified by the regular closed shell interactions. They are predicted to have the typical HB (hydrogen bond) nature with covalency for 1 and 2 but the nature of the molecular complex formation through CT for 3 and 4. The AE–*–BE interactions in models A–D are predicted to be weaker than those in 1–4.
 Please wait while we load your content...
Please wait while we load your content...

