
The enantioselectivity in asymmetric ketone hydrogenation catalyzed by RuH2(diphosphine)(diamine) complexes: insights from a 3D-QSSR and DFT study†
Abstract
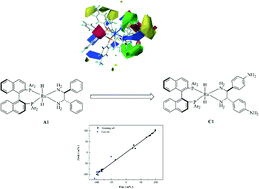
A three-dimensional quantitative structure–selectivity relationship (3D-QSSR) model was developed to investigate the enantioselectivity in asymmetric ketone hydrogenation (AKH) catalyzed by RuH2(diphosphine)(diamine) complexes, through a comparative molecular field analysis (CoMFA). The predicted enantiomeric excess (ee) of the chiral alcohol products was in good agreement with the experimental ones, and the developed model showed good statistics in terms of correlation coefficients (q2 = 0.798, r2 = 0.996). The predictive power of the developed 3D-QSSR model was further proved by a test set of 5 ruthenium complexes, with an r2 of 0.974. The contour map analysis illustrated the sterically and electrostatically favored regions of the ruthenium catalysts for improving the enantioselectivity in the asymmetric hydrogenation. Under the guidance of the model, we modified the structure of the catalyst RuH2[(S)-tolbinap][(S,S)-dpen] (A1) to form the structure RuH2[(S)-tolbinap][(S,S)-dpen-NH2] (C1) where the aromatic rings of the dpen are substituted with amino groups in the para position. The theoretically predicted catalyst, C1, shows a theoretically calculated increase in the ee of AKH by 6.2%. In addition, a computational validation was performed for catalyst C1 under the density function theory (DFT), and a larger calculated difference in energy barriers in the hydrogen transfer step accounted for the enhanced enantioselectivity. In conclusion, the 3D-QSSR method could provide a plausible design criterion for the homogeneous transition-metal (TM) catalysts of asymmetric hydrogenation.
 Please wait while we load your content...
Please wait while we load your content...

