
Towards physical interpretation of substituent effects: the case of meta- and para-substituted anilines†
Abstract
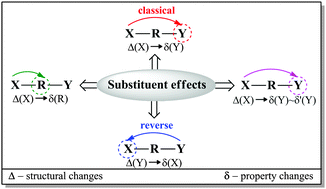
Quantum chemical modeling was used to investigate the electron-donating properties of the amino group in a series of meta- and para-X-substituted anilines (X = NMe2, NH2, OH, OMe, CH3, H, F, Cl, CF3, CN, CHO, COMe, CONH2, COOH, NO2, and NO). Different methods (HF, B3LYP, and M06-2X) and basis sets (6-31+G(d,p), 6-311++G(d,p), and aug-cc-pVDZ) were applied and compared with the MP2 approach. The B3LYP/6-311++G(d,p) method was chosen as the most appropriate one. The substituent properties were described by σ, cSAR(X) and SESE descriptors; the amino group was characterized by structural (dCN, dNH and ΣNH2) and electronic [δ(N) and cSAR(NH2)] parameters; whereas the transmitting moiety was characterized by aromaticity indices HOMA and NICS, as well as by QTAIM characteristics at the ring critical point. All the used parameters were found to be mutually interrelated with much better correlations for the para-derivatives than the meta-derivatives. It was numerically confirmed that sensitivity of the amino group to the substituent effect was greater by over three times when the substituent was located in the para-position. In the case of the meta-derivatives, variability of characteristics for both the reaction center and the substituent was small. The reverse substituent effect was clearly shown by comparison of the cSAR(X) characteristics for monosubstituted benzenes, and meta- and para-substituted anilines.
- This article is part of the themed collection: Electron delocalization and aromaticity: 150 years of the Kekulé benzene structure
 Please wait while we load your content...
Please wait while we load your content...

