
A density functional theory protocol for the calculation of redox potentials of copper complexes†
Abstract
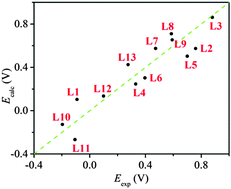
A density functional theory (DFT) protocol for the calculation of redox potentials of copper complexes is developed based on 13 model copper complexes. The redox potentials are calculated in terms of Gibbs free energy change of the redox reaction at the theory level of CAM-B3LYP/6-31+G(d,p)/SMD, with the overall Gibbs free energy change being partitioned into the Gibbs free energy change of the gas phase reaction and the Gibbs free energy change of solvation. In addition, the calculated Gibbs free energy change of solvation is corrected by a unified correction factor of −0.258 eV as the second-layer Gibbs free energy change of solvation and other interactions for each redox reaction. And an empirical Gibbs free energy change of solvation at −0.348 eV is applied to each water molecule if the number of inner-sphere water molecule changes during the redox reaction. Satisfactory agreements between the DFT calculated and experimental results are obtained, with a maximum absolute error at 0.197 V, a mean absolute error at 0.114 V and a standard deviation at 0.133 V. Finally, it is concluded that the accurate prediction of redox potentials is dependent on the accurate prediction of geometrical structures as well as on geometrical conservation during the redox reaction.
 Please wait while we load your content...
Please wait while we load your content...

