
Assay for lignin breakdown based on lignin films: insights into the Fenton reaction with insoluble lignin†
Abstract
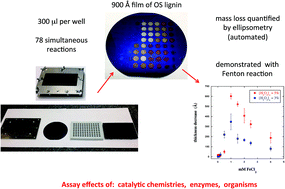
We report a new assay for breakdown of high molecular weight, insoluble lignin based on lignin films. In this method, decrease in film thickness is detected upon solubilization of mass through either chemical alteration of the lignin or molecular weight reduction. The assay was performed with organosolv lignin, the only chemical modification being an oxidative pretreatment to provide film stability with respect to dissolution. The assay is sensitive to release of as little as 20 Å of material from the film. A multiplexed format was developed using a silicone block in the form of a standard 96-well plate, allowing simultaneous assaying of a large number of reaction conditions. The assay was demonstrated using the Fenton reaction, revealing new insights into the physicochemical aspects of this reaction system with insoluble lignin. In particular, mass solubilized from the film was found to pass through a maximum as a function of the initial concentration of FeCl2 ([FeCl2]o), with the maximum occurring at [FeCl2]o = 1 mM for [H2O2]o = 5%. At that condition, solubilization of mass occurs in two stages. The reaction produces mostly ring-opened products of mass greater than 700 g mol−1, along with a minority of low molecular weight aromatics. The new insight from this work is an important step toward optimizing this complex reaction system for effective lignin breakdown.
 Please wait while we load your content...
Please wait while we load your content...

