
Timescales of N–H bond dissociation in pyrrole: a nonadiabatic dynamics study†
Abstract
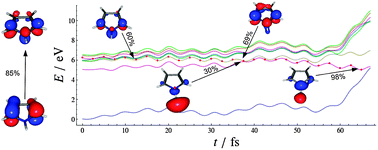
The excitation wavelength dependent photodynamics of pyrrole are investigated by nonadiabatic trajectory-surface-hopping dynamics simulations based on time dependent density functional theory (TDDFT) and the algebraic diagrammatic construction method to the second order (ADC(2)). The ADC(2) results confirm that the N–H bond dissociation occurring upon excitation at the origin of the first excited state, S1(πσ*), is driven by tunnelling [Roberts et al., Faraday Discuss., 2013, 163, 95] as a barrier of ΔE = 1780 cm−1 traps the population in a quasi-bound minimum. Upon excitation to S1(πσ*) in the wavelength range of 236–240 nm, direct dissociation of the N–H bond takes place with a time constant of 28 fs. The computed time constant is in very good agreement with recently reported measurements. Excitation to the lowest B2 state in the 198–202 nm range returns a time constant for N–H fission of 48 fs at the B3LYP/def2-TZVPD level, in perfect agreement with the experiment [Roberts et al. Faraday Discuss., 2013, 163, 95]. For the same wavelength range the ADC(2)/aug-cc-pVDZ decay constant is more than three times longer than the experimentally reported one. The accuracy of the B3LYP/def2-TZVPD dynamics is checked against reference complete-active-space second-order perturbation theory (CASPT2) calculations and explained in terms of correct topography of the ππ* surface and the lack of mixing between the ππ* and the 3px Rydberg states which occurs in the ADC(2) method.
 Please wait while we load your content...
Please wait while we load your content...

