
A new scoring function for protein–protein docking that identifies native structures with unprecedented accuracy†
Abstract
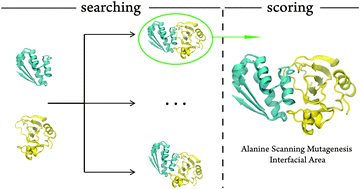
Protein–protein (P–P) 3D structures are fundamental to structural biology and drug discovery. However, most of them have never been determined. Many docking algorithms were developed for that purpose, but they have a very limited accuracy in generating native-like structures and identifying the most correct one, in particular when a single answer is asked for. With such a low success rate it is difficult to point out one docked structure as being native-like. Here we present a new, high accuracy, scoring method to identify the 3D structure of P–P complexes among a set of trial poses. It incorporates alanine scanning mutagenesis experimental data that need to be obtained a priori. The scoring scheme works by matching the computational and the experimental alanine scanning mutagenesis results. The size of the trial P–P interface area is also taken into account. We show that the method ranks the trial structures and identifies the native-like structures with unprecedented accuracy (∼94%), providing the correct P–P 3D structures that biochemists and molecular biologists need to pursue their studies. With such a success rate, the bottleneck of protein–protein docking moves from the scoring to searching algorithms.
 Please wait while we load your content...
Please wait while we load your content...

