
Thermodynamic insights into the self-assembly of capped nanoparticles using molecular dynamic simulations†
Abstract
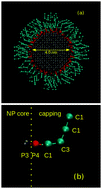
Although the molecular modeling of self-assembling processes stands as a challenging research issue, there have been a number of breakthroughs in recent years. This report describes the use of large-scale molecular dynamics simulations with coarse grained models to study the spontaneous self-assembling of capped nanoparticles in chloroform suspension. A model system comprising 125 nanoparticles in chloroform evolved spontaneously from a regular array of independent nanoparticles to a single thread-like, ramified superstructure spanning the whole simulation box. The aggregation process proceeded by means of two complementary mechanisms, the first characterized by reactive collisions between monomers and oligomers, which were permanently trapped into the growing superstructure, and the second a slow structural reorganization of the nanoparticle packing. Altogether, these aggregation processes were over after ca. 0.6 μs and the system remained structurally and energetically stable until 1 μs. The thread-like structure closely resembles the TEM images of capped ZrO2, but a better comparison with experimental results was obtained by the deposition of the suspension over a graphene solid substrate, followed by the complete solvent evaporation. The agreement between the main structural features from this simulation and those from the TEM experiment was excellent and validated the model system. In order to shed further light on the origins of the stable aggregation of the nanoparticles, the Gibbs energy of aggregation was computed, along with its enthalpy and entropy contributions, both in chloroform and in a vacuum. The thermodynamic parameters arising from the modeling are consistent with larger nanoparticles in chloroform due to the solvent-swelled organic layer and the overall effect of the solvent was the partial destabilization of the aggregated state as compared to the vacuum system. The modeling strategy has been proved effective and reliable to describe the self-assembling of capped nanoparticles, but we must acknowledge the fact that larger model systems and longer timescales will be necessary in future investigations in order to assess structural and dynamical information approaching the behavior of macroscopic systems.
 Please wait while we load your content...
Please wait while we load your content...

