
Effect of processing conditions on the crystallinity and structure of carbonated calcium hydroxyapatite (CHAp)
Abstract
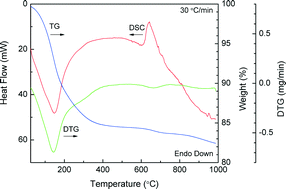
Previous synthesis routes created apatites in low crystallinity and high crystallinity states, but a wider range will extend the design capabilities of apatites for hard tissue replacements. While high crystallinity apatites are more conventional, this work investigated lower crystallinity variations from an amorphous state to low crystallinity apatite. Carbonated hydroxyapatite was prepared by precipitating an amorphous phase followed by crystallization at 650 °C at slow (5 °C min−1) and fast heating rates (60 °C min−1). The effect of processing conditions on crystallinity and structural changes was evaluated by thermal analysis, X-ray diffraction, transmission electron microscopy, Fourier transform infrared and Raman spectroscopy. Furthermore, peak deconvolution of IR and Raman spectra resolved the carbonate and phosphate bands and revealed the carbonate and crystalline phase content in CHAp. Similar to precipitation of crystalline apatites, the crystallization at elevated temperature led to carbonate in both the phosphate and hydroxyl positions. Heating at 650 °C provided a nanosized spherical hydroxyapatite containing carbonate controlled by the heating rate. This creates a mechanism for creating a large range in crystallinity with a greater resorption capability for regenerative medicine.
 Please wait while we load your content...
Please wait while we load your content...

