
Visible-light radical reaction designed by Ru- and Ir-based photoredox catalysis
Abstract
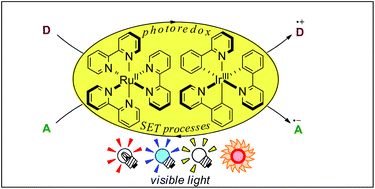
Photoredox catalysis by well-known ruthenium(II) polypyridine complexes and the relevant Ir cyclometalated derivatives has become a powerful tool for redox reactions in synthetic organic chemistry, because they can effectively catalyze single-electron-transfer (SET) processes by irradiation with visible light. Remarkably, since 2008, this photocatalytic system has gained importance in radical reactions from the viewpoint of not only a useful and selective protocol but also green chemistry. In this review, we will describe recent developments of radical reactions involving various carbon-centered radicals through photoredox processes mediated by Ru- and Ir-based photocatalysts.
- This article is part of the themed collections: Inorganic Chemistry Frontiers Outstanding Paper Awards 2014-2023 and 2014 Inorganic Chemistry Frontiers Review Articles
 Please wait while we load your content...
Please wait while we load your content...

