
A theoretical study of ternary indole–cation–anion complexes†
Abstract
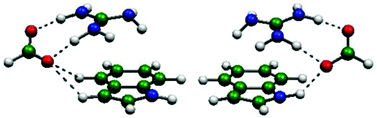
The simultaneous interactions of an anion and a cation with a π system were investigated by MP2 and M06-2X theoretical calculations. Indole was chosen as a model π system for its relevance in biological environments. Two different orientations of the anion, interacting with the N–H and with the C–H groups of indole, were considered. The four cations (Na+, NH4+, C(NH2)3+ and N(CH3)4+) and the four anions (Cl−, NO3−, HCOO− and BF4−) included in the study are of biological interest. The total interaction energy of the ternary complexes was calculated and separated into its two- and three-body components and all of them are further divided into their electrostatic, exchange, repulsion, polarization and dispersion contributions using the local molecular orbital-energy decomposition analysis (LMO-EDA) methodology. The binding energy of the indole-cation-anion complexes depends on both ions, with the cation having the strongest effect. The intense cation–anion attraction determines the geometric and energetic features in all ternary complexes. These structures, with both ions on the same side of the π system, show an anti-cooperative interaction. However, the interaction is not only determined by electrostatics, but also the polarization contribution is important. Specific interactions like the one established between the anion and the N–H group of indole or the proton transfer between an acidic cation and a basic anion play a significant role in the energetics and the structure of particular complexes. The presence of the polar solvent as modelled with the polarizable continuum model (PCM) does not seem to have a significant effect on the geometry of the ternary complexes, but drastically weakens the interaction energy. Also, the strength of the interaction is reduced at a faster rate when the anion is pushed away, compared to the results obtained in the gas phase. The combination of PCM with the addition of one water molecule indicates that the PCM method properly reproduces the main energetic and geometrical changes, even at the quantitative level, but the explicit hydration allows refining the solvent effect and detecting cases that do not follow the general trend.
 Please wait while we load your content...
Please wait while we load your content...

