
The asymmetric syntheses of pyrrolizidines, indolizidines and quinolizidines via two sequential tandem ring-closure/N-debenzylation processes†
Abstract
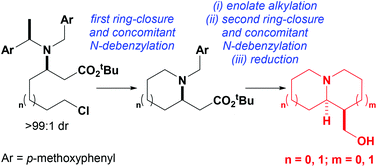
Concise asymmetric syntheses of (−)-lupinine, (+)-isoretronecanol, (+)-5-epi-tashiromine and (R,R)-1-(hydroxymethyl)octahydroindolizine (the azabicyclic core within stellettamides A–C) have been achieved in 8 steps or fewer from commercially available starting materials. The key steps in these syntheses involved the preparation of enantiopure β-amino esters, upon conjugate addition of lithium (R)-N-(p-methoxybenzyl)-N-(α-methyl-p-methoxybenzyl)amide to either ζ-chloro or ζ-hydroxy substituted tert-butyl (E)-hept-2-enoate, or ε-chloro or ε-hydroxy substituted tert-butyl (E)-hex-2-enoate. Activation of the ω-substituent as a leaving group led to SN2-type ring-closure, which occurred with concomitant N-debenzylation via an E1-type deprotection step, to give the corresponding pyrrolidine or piperidine in good yield. Subsequent alkylation of these enantiopure azacycles, followed by a second ring-closure/concomitant N-debenzylation step formed the pyrrolizidine, indolizidine or quinolizidine motif, and reduction with LiAlH4 gave the target compounds in diastereoisomerically and enantiomerically pure form.
 Please wait while we load your content...
Please wait while we load your content...

