
Bisactinyl halogenated complexes: relativistic density functional theory calculation and experimental synthesis†
Abstract
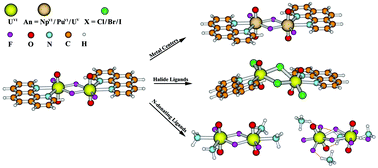
Relativistic density functional theory was used to explore a series of bisactinyl complexes, [(phen)(AnVIO2)(μ2-F)(F)]2 (An = U (1), Np (2) and Pu (3); phen = ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O indicate the partial triple bonding character, and agree with the trends in the AnO stretching vibrational frequencies. The free energies of formation reactions of 1–11 calculated in the aqueous solution demonstrate that 1, 8 and 11 are thermodynamically stable. In this work, we have successfully synthesized 1 and [Me2NH2]4[(F)2(UVIO2)(μ2-F)(F)]2 (12), which is represented by theoretical model complex 11. Their characterizations of the single crystal X-ray diffraction and infrared are consistent with the calculated results; the measured fine-structured fluorescent emissions were assigned as transitions from the UO bonding to the U (f) orbital by analyzing the electronic structure and
O indicate the partial triple bonding character, and agree with the trends in the AnO stretching vibrational frequencies. The free energies of formation reactions of 1–11 calculated in the aqueous solution demonstrate that 1, 8 and 11 are thermodynamically stable. In this work, we have successfully synthesized 1 and [Me2NH2]4[(F)2(UVIO2)(μ2-F)(F)]2 (12), which is represented by theoretical model complex 11. Their characterizations of the single crystal X-ray diffraction and infrared are consistent with the calculated results; the measured fine-structured fluorescent emissions were assigned as transitions from the UO bonding to the U (f) orbital by analyzing the electronic structure and
 Please wait while we load your content...
Please wait while we load your content...

