
Subnanometer-sized Pt/Sn alloy cluster catalysts for the dehydrogenation of linear alkanes
Abstract
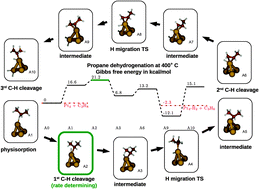
The reaction pathways for the dehydrogenation of ethane, propane, and butane, over Pt are analyzed using density functional theory (DFT). Pt nanoparticles are represented by a tetrahedral Pt4 cluster. The objectives of this work were to establish which step is rate limiting and which one controls the selectivity for forming alkenes as opposed to causing further dehydrogenation of adsorbed alkenes to produce precursors responsible for catalyst deactivation due to coking. Further objectives of this work are to identify the role of adsorbed hydrogen, derived from H2 fed together with the alkane, on the reaction pathway, and the role of replacing one of the four Pt atoms by a Sn atom. A comparison of Gibbs free energies shows that in all cases the rate-determining step is cleavage of a C–H bond upon alkane adsorption. The selectivity to alkene formation versus precursors to coking is dictated by the relative magnitudes of the activation energies for alkene desorption and dehydrogenation of the adsorbed alkene. The presence of an adsorbed H atom on the cluster facilitates alkene desorption relative to dehydrogenation of the adsorbed alkene. Substitution of a Sn atom in the cluster to produce a Pt3Sn cluster leads to a downward shift of the potential energy surface for the reaction and causes an increase of the activity of the catalyst as suggested by recent experiments due to the lower net activation barrier for the rate limiting step. However, the introduction of Sn does not alter the relative activation barriers for gas-phase alkene formation versus loss of hydrogen from the adsorbed alkene, the process leading to the formation of coke precursors.
 Please wait while we load your content...
Please wait while we load your content...

