
Fast and accurate computational modeling of adsorption on graphene: a dispersion interaction challenge†
Abstract
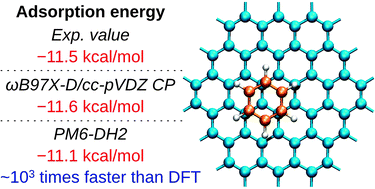
Understanding molecular interactions of graphene is a question of key importance to design new materials and catalytic systems for practical usage. Although for small models good accuracy was demonstrated in theoretical analysis with ab initio and density functional methods, the application to real-size systems with thousands of atoms is currently hardly possible on routine bases due to the high computational cost. In the present study we report that incorporation of dispersion correction led to the principal improvement in the description of graphene systems at a semi-empirical level. The accuracy and the scope of the calculations were explored for a wide range of molecules adsorbed on graphene surfaces (H2, N2, CO, CO2, NH3, CH4, H2O, benzene, naphthalene, coronene, ovalene and cyclohexane). As a challenging parameter, the calculated adsorption energy of aromatic hydrocarbons on graphene Eads = −1.8 ± 0.1 kcal mol−1 (per one carbon atom) at the PM6-DH2 level was in excellent agreement with the experimentally determined value of Eads = −1.7 ± 0.3 kcal mol−1. The dispersion corrected semi-empirical method was found to be a remarkable computational tool suitable for everyday laboratory studies of real-size graphene systems. Significant performance improvement (ca. 103 times faster) and excellent accuracy were found as compared to the ωB97X-D density functional calculations.
 Please wait while we load your content...
Please wait while we load your content...

