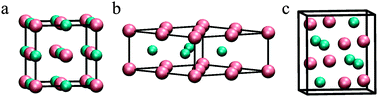
The geometric and electronic structure of catalytically relevant molybdenum carbide phases (cubic δ-MoC, hexagonal α-MoC, and orthorhombic β-Mo2C) and their low Miller-index surfaces have been investigated by means of periodic density functional theory (DFT) based calculations with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional. Comparison to available experimental data indicates that this functional is particularly well suited to study these materials. The calculations reveal that β-Mo2C has a stronger metallic character than the other two polymorphs, both β-Mo2C and δ-MoC have a large ionic contribution, and δ- and α-MoC exhibit the strongest covalent character. Among the various surfaces explored, the calculations reveal the high stability of the δ-MoC(001) nonpolar surface, Mo- and C-terminated (001) polar surfaces of α-MoC, and the nonpolar (011) surface of β-Mo2C. A substantially low work function of only 3.4 eV is predicted for β-Mo2C(011), suggesting that this system is particularly well suited for (electro)catalytic processes where surface → adsorbate electron transfer is essential. The overall implications for heterogeneously catalysed reactions by these molybdenum carbide nanoparticles are also discussed.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


