
Donor and acceptor levels of organic photovoltaic compounds from first principles
Abstract
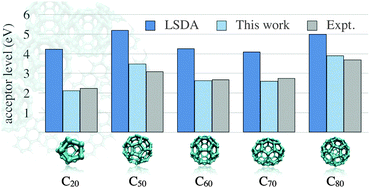
Accurate and efficient approaches to predict the optical properties of organic semiconducting compounds could accelerate the search for efficient organic photovoltaic materials. Nevertheless, predicting the optical properties of organic semiconductors has been plagued by the inaccuracy or computational cost of conventional first-principles calculations. In this work, we demonstrate that orbital-dependent density-functional theory based upon Koopmans' condition [Phys. Rev. B, 2010, 82, 115121] is apt for describing donor and acceptor levels for a wide variety of
- This article is part of the themed collection: Charge transfer: experiment, theory and computation
 Please wait while we load your content...
Please wait while we load your content...

