
Defect chemistry of a BaZrO3 Σ3 (111) grain boundary by first principles calculations and space–charge theory
Abstract
Defect calculations from density functional theory are implemented with space–charge theory models to describe the equilibrium defect chemistry of a Σ3 (111) symmetric tilt boundary in BaZrO3. As such, the space–charge potential and the concentrations of 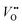 ,
, 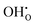 ,
, 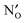 , NH×O and
, NH×O and 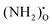 in the bulk, core and space–charge regions of the interface are calculated as a function of temperature and atmospheric conditions. Our results show that the core will be predominated by
in the bulk, core and space–charge regions of the interface are calculated as a function of temperature and atmospheric conditions. Our results show that the core will be predominated by 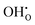 under hydrating conditions and that the space–charge potential increases with
under hydrating conditions and that the space–charge potential increases with 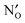 , NH×O and
, NH×O and 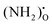 will predominate the core in different temperature regimes and effects of these defects on the space–charge properties are discussed.
will predominate the core in different temperature regimes and effects of these defects on the space–charge properties are discussed.

 Please wait while we load your content...
Please wait while we load your content...

