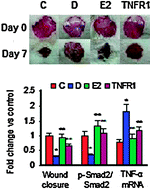
Wound healing (WH) impairment is a well-documented phenomenon in clinical and experimental diabetes. Sex hormones, in addition to a number of signaling pathways including transforming growth factor-β1 (TGF-β1)/Smads and TNF-α/NF-κB in macrophages and fibroblasts, appear to play a cardinal role in determining the rate and nature of WH. We hypothesized that a defect in resolution of inflammation and an enhancement in TNF-α/NF-κB activity induced by estrogen deficiency contribute to the impairment of TGF-β signaling and delayed WH in diabetes models. Goto-Kakizaki (GK) rats and full thickness excisional wounds were used as models for type 2 diabetes (T2D) and WH, respectively. Parameters related to the various stages of WH were assessed using histomorphometry, western blotting, real-time PCR, immunofluorescence microscopy and ELISA-based assays. Retarded re-epithelialization, suppressed angiogenesis, delayed wound closure, reduced estrogen level and heightened states of oxidative stress were characteristic features of T2D wounds. These abnormalities were associated with a defect in resolution of inflammation, shifts in macrophage phenotypes, increased β3-integrin expression, impaired wound TGF-β1 signaling (↓p-Smad2/↑Smad7) and enhanced TNF-α/NFκB activity. Human/rat dermal fibroblasts of T2D, compared to corresponding control values, displayed resistance to TGF-β-mediated responses including cell migration, myofibroblast formation and p-Smad2 generation. A pegylated form of soluble TNF receptor-1 (PEG-sTNF-RI) or estrogen replacement therapy significantly improved re-epithelialization and wound contraction, enhanced TGFβ/Smad signaling, and polarized the differentiation of macrophages toward an M2 or “alternatively” activated phenotype, while limiting secondary inflammatory-mediated injury. Our data suggest that reduced estrogen levels and enhanced TNF-α/NF-κB activity delayed WH in T2D by attenuating TGFβ/Smad signaling and impairing the resolution of inflammation; most of these defects were ameliorated with estrogen and/or PEG-sTNF-RI therapy.

 Please wait while we load your content...
Please wait while we load your content...