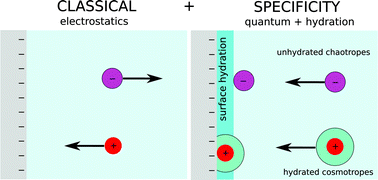
The classical Derjaguin–Landau–Verwey–Overbeek (DLVO) theory of colloids, and corresponding theories of electrolytes, are unable to explain ion specific forces between colloidal particles quantitatively. The same is true generally, for surfactant aggregates, lipids, proteins, for zeta and membrane potentials and in adsorption phenomena. Even with fitting parameters the theory is not predictive. The classical theories of interactions begin with continuum solvent electrostatic (double layer) forces. Extensions to include surface hydration are taken care of with concepts like inner and outer Helmholtz planes, and “dressed” ion sizes. The opposing quantum mechanical attractive forces (variously termed van der Waals, Hamaker, Lifshitz, dispersion, nonelectrostatic forces) are treated separately from electrostatic forces. The ansatz that separates electrostatic and quantum forces can be shown to be thermodynamically inconsistent. Hofmeister or specific ion effects usually show up above ≈10−2 molar salt. Parameters to accommodate these in terms of hydration and ion size had to be invoked, specific to each case. Ionic dispersion forces, between ions and solvent, for ion–ion and ion–surface interactions are not explicit in classical theories that use “effective” potentials. It can be shown that the missing ionic quantum fluctuation forces have a large role to play in specific ion effects, and in hydration. In a consistent predictive theory they have to be included at the same level as the nonlinear electrostatic forces that form the skeletal framework of standard theory. This poses a challenge. The challenges go further than academic theory and have implications for the interpretation and meaning of concepts like pH, buffers and membrane potentials, and for their experimental interpretation. In this article we overview recent quantitative developments in our evolving understanding of the theoretical origins of specific ion, or Hofmeister effects. These are demonstrated through an analysis that incorporates nonelectrostatic ion–surface and ion–ion dispersion interactions. This is based on ab initio ionic polarisabilities, and finite ion sizes quantified through recent ab initio work. We underline the central role of ionic polarisabilities and of ion size in the nonelectrostatic interactions that involve ions, solvent molecules and interfaces. Examples of mechanisms through which they operate are discussed in detail. An ab initio hydration model that accounts for polarisabilities of the tightly held hydration shell of “cosmotropic” ions is introduced. It is shown how Hofmeister effects depend on an interplay between specific surface chemistry, surface charge density, pH, buffer, and counterion with polarisabilities and ion size. We also discuss how the most recent theories on surface hydration combined with hydrated nonelectrostatic potentials may predict experimental zeta potentials and hydration forces.

 Please wait while we load your content...
Please wait while we load your content...

