
Accuracy of the microsolvation–continuum approach in computing the pKa and the free energies of formation of phosphate species in aqueous solution†
Abstract
First principles density functional theory (Perdew–Burke–Ernzerhof) calculations have been used to compute the hydration properties, aqueous-phase acid dissociation constants (pKa) and Gibbs free energies of formation of small polyphosphates in aqueous solution. The effect of the hydrated environment has been simulated through a hybrid microsolvation–continuum approach, where the 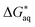 of
of 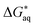 can vary by tenths of kcal mol−1, depending on the particular choice of nH2O for the monomeric and dimeric species. We discuss a methodology for the determination of nH2O; for the orthophosphates the “incremental” binding energy approach is used to determine nH2O, whereas for the polyphosphates the number of explicit
can vary by tenths of kcal mol−1, depending on the particular choice of nH2O for the monomeric and dimeric species. We discuss a methodology for the determination of nH2O; for the orthophosphates the “incremental” binding energy approach is used to determine nH2O, whereas for the polyphosphates the number of explicit

 Please wait while we load your content...
Please wait while we load your content...

