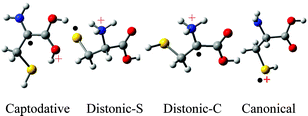
A theoretical study on the structures, relative energies, isomerization reactions and fragmentation pathways of the cysteine radical cation, [NH2CH(CH2SH)COOH]˙+, is reported. Hybrid density functional theory (B3LYP) has been used in conjunction with the 6-311++G(d,p) basis set. The isomer at the global minimum, Captodative-1, has the structure NH2C˙(CH2SH)C(OH)2+; the stability of this ion is attributed to the captodative effect in which the NH2 functions as a powerful π-electron donor and C(OH)2+ as a powerful π-electron acceptor. Ion Distonic-S-1, H3N+CH(CH2S˙)COOH, in which the radical is formally situated on the S atom, is higher in enthalpy (ΔH°0) than Captodative-1 by 6.1 kcal mol–1, but is lower in enthalpy than another isomer Distonic-C-1, H3N+C˙(CH2SH)COOH, by 8.2 kcal mol–1. Isomerization of the canonical radical cation of cysteine, [H2NCH(CH2SH)COOH]˙+, (Canonical-1), to Captodative-1 has an enthalpy of activation of 25.8 kcal mol–1, while the barrier against isomerization of Canonical-1 to Distonic-S-1 is only 9.6 kcal mol–1. Two additional transient tautomers, one with the radical located at Cα and the charge on SH2, and the other a carboxy radical with the charge on NH3, are reported. Plausible fragmentation pathways (losses of small molecules, CO2, CH2S, H2S and NH3, and neutral radicals COOH˙, HSCH2˙ and NH2˙) from Canonical-1 are examined.

 Please wait while we load your content...
Please wait while we load your content...

