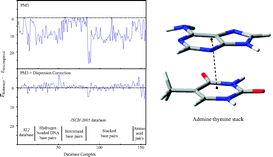
Semi-empirical calculations including an empirical dispersive correction are used to calculate intermolecular interaction energies and structures for a large database containing 156 biologically relevant molecules (hydrogen-bonded DNA base pairs, interstrand base pairs, stacked base pairs and amino acid base pairs) for which MP2 and CCSD(T) complete basis set (CBS) limit estimates of the interaction energies are available. The dispersion corrected semi-empirical methods are parameterised against a small training set of 22 complexes having a range of biologically important non-covalent interactions. For the full molecule set (156 complexes), compared to the high-level ab initio database, the mean unsigned errors of the interaction energies at the corrected semi-empirical level are 1.1 (AM1-D) and 1.2 (PM3-D) kcal mol−1, being a significant improvement over existing AM1 and PM3 methods (8.6 and 8.2 kcal mol−1). Importantly, the new semi-empirical methods are capable of describing the diverse range of biological interactions, most notably stacking interactions, which are poorly described by both current AM1 and PM3 methods and by many DFT functionals. The new methods require no more computer time than existing semi-empirical methods and therefore represent an important advance in the study of important biological interactions.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


