
Direct quantum dynamics using variational multi-configuration Gaussian wavepackets. Implementation details and test case
Abstract
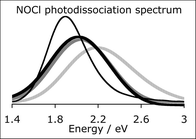
We present here a direct quantum dynamics method using variational multi-configuration Gaussian wavepackets. Based on the efficient multi-configuration time-dependent Hartree wavepacket propagation algorithm, it uses on-the-fly quantum chemical calculation of the potential energy and its derivatives rather than fitted surfaces. Intermediate results are stored in a database so that expensive quantum chemical computations can be recycled. This method is intended to treat quantum effects in the photochemistry of large molecules and the use of Cartesian coordinates to perform direct dynamics is discussed with a comparison between Cartesian coordinates of Jacobi vectors and Cartesian coordinates of the nuclei, using various free and constrained approaches depending on the way the rotation is treated. As a test calculation to be compared to full quantum dynamics it is applied here to the computation of the photodissociation spectrum of
 Please wait while we load your content...
Please wait while we load your content...

